
Abstract
Vector-borne microbes are subject to the ecological constraints of two distinct microenvironments: that in the arthropod vector and that in the blood of its vertebrate host. Because the structure of bacterial communities in these two microenvironments may substantially affect the abundance of vector-borne microbes, it is important to understand the relationship between bacterial communities in both microenvironments and the determinants that shape them. We used pyrosequencing analyses to compare the structure of bacterial communities in Synosternus cleopatrae fleas and in the blood of their Gerbillus andersoni hosts. We also monitored the interindividual and seasonal variability in these bacterial communities by sampling the same individual wild rodents during the spring and again during the summer. We show that the bacterial communities in each sample type (blood, female flea or male flea) had a similar phylotype composition among host individuals, but exhibited seasonal variability that was not directly associated with host characteristics. The structure of bacterial communities in male fleas and in the blood of their rodent hosts was remarkably similar and was dominated by flea-borne Bartonella and Mycoplasma phylotypes. A lower abundance of flea-borne bacteria and the presence of Wolbachia phylotypes distinguished bacterial communities in female fleas from those in male fleas and in rodent blood. These results suggest that the overall abundance of a certain vector-borne microbe is more likely to be determined by the abundance of endosymbiotic bacteria in the vector, abundance of other vector-borne microbes co-occurring in the vector and in the host blood and by seasonal changes, than by host characteristics.
Similar content being viewed by others

Introduction
The identification of potential ecological factors that affect the abundance of vector-borne microbes could have a marked impact on our ability to prevent and control vector-borne diseases (Lemon et al., 2008). Importantly, vector-borne microbes are subject to ecological conditions and constraints in two different microenvironments: that in the arthropod vector and that in the blood of its vertebrate host. One important factor that affects these conditions—and therefore the microenvironment that vector-borne microbes are subject to—is the resident bacterial communities, the members of which can have different relationships with the host and vector organisms, ranging from mutualistic to commensalistic or parasitic (Clay et al., 2006). In addition, interactions between different microbes within each microenvironment may affect both the density of a certain vector-borne microbe and its transmission rate and virulence to human hosts (Pumpuni et al., 1993; Lipsitch et al., 1996; Macaluso et al., 2002; de la Fuente et al., 2003; Gonzalez-Ceron et al., 2003; Lively et al., 2005; Telfer et al., 2010; Cirimotich et al., 2011). Thus, characterizing the structure of bacterial communities (that is, richness, diversity and composition of phylotypes) in both the vector and the host, and the factors that shape them, is important for understanding the determinants of the overall abundance of vector-borne microbes.
Most studies of vector-borne microbiota have focused on microbial communities within the microenvironment of the arthropod vector, including studies on lice (Reed and Hafner, 2002), fleas (Jones et al., 2010; Hawlena et al., 2013), mosquitoes (Rani et al., 2009; Osei-Poku et al., 2012), flies (Hornok et al., 2008), ticks (Heise et al., 2010; Andreotti et al., 2011; Hawlena et al., 2013), mites (Netusil et al., 2005; Reeves et al., 2006) and leeches (Worthen et al., 2006). Diverse microbial communities were found in these vectors, and most microbial species were represented sparsely or occurred in only a few individual vectors. Some studies also indicated that the composition of these microbial communities often varies across time and space (Kirstein et al., 1997; Wielinga et al., 2006; van Overbeek et al., 2008; Jones et al., 2010) and that these variations are linked largely to the characteristics of the arthropod (for example, its species, life stage, diet and blood engorgement level; Pidiyar et al., 2004; Moreno et al., 2006; Heise et al., 2010; Jones et al., 2010; Hawlena et al., 2013). Nevertheless, because vector-borne microbes spend a part of their lives in the blood of the vertebrate host, it is important to consider also the internal environment of this host. For instance, microbes in the blood—similar to other microbes in the vertebrate gut, secretion glands, plumage, cloaca and skin—may be influenced by host species, diet, personality, body mass, sex, age and health status (see, for example, Alexy et al., 2003; Voigt et al., 2005; Fierer et al., 2008; Turnbaugh et al., 2009; Martin et al., 2010; Koenig et al., 2011; Muegge et al., 2011; Roeselers et al., 2011; Saag et al., 2011; Yatsunenko et al., 2012; Faith et al., 2013; van Dongen et al., 2013; Gavish et al., 2014; Kedem et al., 2014; Kueneman et al., 2014; Leclaire et al., 2014).
Here, we therefore address two main questions. (1) To what extent is the structure of bacterial communities in arthropod vectors similar to that in the blood of their vertebrate host? (2) What are the most important determinants of the structure of the two communities, and to what extent are they stable between host individuals and over time? To answer these questions, we characterized the structure of bacterial communities in bloodsucking arthropod vectors (fleas) and in the blood of their vertebrate hosts (wild gerbils). Female fleas, male fleas and blood samples were obtained from the same gerbils at two different time points: first during the spring, when some individuals were juveniles and others were adults, and then ∼4 months later, at the end of the summer, when all individuals were adults. We hypothesized that the structure of bacterial communities in fleas would differ from those in blood samples because the blood may contain directly transmitted bacteria and phylotypes that are transmitted by non-flea vectors (for example, helminths and ticks; Hawlena et al., 2006 and Hawlena H, unpublished data) in addition to the flea-borne bacteria (that should appear in both communities). We also hypothesized that the bacterial communities in fleas would show greater phylotype richness and diversity than those in the blood samples, because fleas may harbor both bacterial endosymbionts and bacteria from the very rich soil microbiome (that is, bacteria originally acquired during the larval stage of the fleas and then transmitted transstadially to adult fleas; Torsvik et al., 1990). In addition, although the bacterial communities in the gerbils and fleas are largely influenced by the internal environment of the gerbil and flea, respectively, bacterial communities in the fleas are also influenced by the external environment of the gerbil. Hence, we also expected that the stability and determinants of bacterial community structure in the blood samples would show a higher interindividual variability, a lower seasonal variability and a stronger dependency on host characteristics than those in the fleas.
Materials and methods
Study design
We trapped and identified juvenile and adult gerbils (Gerbillus andersoni) and obtained their fleas (Synosternus cleopatrae) and blood samples on two occasions: (1) during spring (April–May), which is the reproductive season of the gerbils, and (2) during the end of summer (September). At the latter sampling period, all individuals were adults. We quantified changes in the structure of bacterial communities in the flea vectors and in the blood of their rodent hosts in relation to the host characteristics (individual identity, age and sex), sample type (female flea, male flea or rodent blood) and sampling season (spring or summer). Because the same individuals were captured in two sampling seasons, this design enabled differentiating between effects related to vector or host characteristics and effects related to temporal (seasonal) changes that occur in all host individuals. In addition, our sampling of individuals belonging to the same species, occupying the same macrohabitat (semistabilized sandy dunes; Abramsky et al., 1985) and feeding on a similar diet (composed mainly of seeds; Bar et al., 1981) deflates the type II error. Thus, our experimental design increased the likelihood of observing age- and sex-related effects in bacterial communities and avoided sampling challenges common to studies in humans or in laboratory specimens.
Rodent capture, tagging and sampling
We trapped rodents in each of ten 1 ha plots located in the Western Negev, Israel (see Supplementary Methods). We tagged each individual and determined its species, sex, weight, age group (juveniles <18 g; adults >18 g, following Hawlena et al., 2006) and, in female rodents, the reproductive status. We collected all fleas (see Supplementary Methods). In addition, using sterile capillaries, we drew 100–200 μl of blood from the retro-orbital sinus of each rodent and stored the blood in EDTA blood collection tubes at −20 °C until DNA extraction. Because of the sensitivity of the eyes, we could not sterilize the local area before drawing the blood. Therefore, to minimize contamination, we performed the bleeding process as quickly as possible. In addition, we always changed capillaries when we had to stab the retro-orbital sinus of a given individual more than once. Such efforts cannot completely prevent surface contamination, and, as a result, some bacterial phylotypes associated with the skin or eye of the rodent could have been artificially added to the pool of detected phylotypes. However, from the 40 phylotypes that we detected, only five represent genera that are associated with the skin or eye microbiomes of rodents (that is, Staphylococcus spp. and Streptococcus spp.; Cullen, 2003; Coster et al., 2008; Montiani-Ferreira et al., 2008; Scharschmidt et al., 2009; Meekins et al., 2014). From these five phylotypes, three were each confined to one sample, one was detected in four samples and one was detected in five samples. Hence, such a contamination, if occurred, had a negligible effect on our results. Moreover, as all host individuals were subjected to exactly the same sampling procedure, possible surface contaminants should have only increased the number of bacterial phylotypes that are not associated with the flea vectors and increase the variance among bacterial communities in the blood of different host individuals but not across seasons.
The trapping and handling protocol was approved by the Committee for the Ethical Care and Use of Animals in Experiments of Ben-Gurion University of the Negev (permission no. IL-14-03-2011) and by the Nature and National Parks Protection Authority (permission no. 2011/38146).
Characterization of bacterial communities
Fleas were first surface sterilized by washing them thoroughly three times in 70% ethanol (Hawlena et al., 2013). Although this simple method could not completely prevent surface contamination, it allowed us to remove most contaminants without damaging the quality of the DNA (H Hawlena et al., unpublished data). Indeed, of the 40 phylotypes that we have detected, only two phylotypes—Azovibrio sp. and Saccharothrix sp.—are possible contaminants; the rest of the phylotypes that we found in fleas represent genera that are known as mutualists of arthropods or that are present in the digestive system of arthropods. As each of these candidate contaminants was found in only a single sample, we believe that their presence should have a negligible influence on our findings.
To extract DNA from fleas, we used the DNeasy Blood and Tissue Kit (QIAGEN, Valencia, CA, USA) according to the QIAGEN ‘purification of total DNA from ticks’ supplementary protocol for the detection of Borrelia DNA (Hawlena et al., 2013). To extract DNA from rodent blood samples, we used a Bacteremia DNA isolation kit (MoBio Laboratories, Carlsbad, CA, USA). We added 50 μl blood to the Microbead tube and followed the manufacturer’s instructions. We added a negative control to each extraction session by adding all reagents to phosphate-buffered saline instead of fleas or blood. We subjected the DNA extracts and nine negative controls to V1–V3 region 16S rRNA PCR, following Hawlena et al. (2013), and performed 454 pyrosequencing on a Roche/454 GS-FLX Titanium system (Branford, CT, USA).
We processed the pyrosequencing reads with Mothur (version 1.22.0), as described in Schloss et al. (2011). Briefly, we assigned bacterial sequences to specific flea or blood samples only if they perfectly matched the primer barcode sequence. We trimmed the primer and barcode sequences from the remaining sequences, trimmed sequences <200 bp. We performed an additional denoise step to remove low-quality sequences, followed by chimera detection and elimination.
We binned the sequences into phylotypes, where a phylotype was defined as a group of microbes that are commonly characterized by the level of sequence similarity between small subunit (16S) rRNA genes. We grouped two sequences into a single ‘species’-level phylotype if they were ≥97% identical over their overlap regions (an equivalent for the species level in macroorganisms; Stackebrandt and Goebel, 1994). We classified the phylotypes to the genus level by using the classifier function embedded in the Mothur package, where 60% RDP (Ribosomal Database Project) classifier confidence cutoffs were applied. To confirm the classifications, we also randomly sampled 100 member sequences for each phylotype and compared them with the RDP 10.26 database by using BlastN (Altschul et al., 1997) with an E-value cutoff of 1E−20 (Maidak et al., 1996). We included in the analyses only phylotypes that had a significantly higher relative abundance (≥2% of the bacterial community in any one sample) in the flea or blood samples than in the negative controls, following Jones et al. (2010). All sequences and the metadata file are available from the SRA (Sequence Read Archive) database (study accession SRP049346).
Data analysis
Details of the data analysis procedures are given in the Supplementary Methods section and are shown here in brief. All analyses were performed at the phylotype level. Specifically, the dependent variables in this study were bacterial community composition (binary Bray–Curtis), richness (number of phylotypes in a sample) and diversity (community richness and evenness), and the independent variables were host characteristics, sample type, sampling season and their second and third interaction terms.
To analyze the effects of the independent variables on community composition, we performed a nonparametric multivariate multiple regression analysis with a stepwise forward-selection procedure. Community composition was assessed based on the Bray–Curtis dissimilarity index (Magurran, 1988) as follows: first, relative abundances were fourth-root transformed, such that highly abundant phylotypes did not dominate the analyses; then, the ‘phylotype-sample’ abundance matrix was converted to a ‘sample-sample’ distance matrix, by using PRIMER-E (Clarke, 1993).
We determined the overall contribution of individual phylotypes within a community to the observed differences by using SIMPER, an analysis tool within the PRIMER-E software, and visualized patterns in multivariate data with an unconstrained nonmetric multidimensional scaling plot. We analyzed associations between the independent and the other two dependent variables by using the model selection approach (Burnham and Anderson, 2002; see Table 1 and Supplementary Methods for details). We based all models on generalized estimating equations for the repeated analysis of individuals across time, with a gamma distribution for the diversity data and a negative binomial distribution for the richness data. Because the dependent variables may have potentially been affected by the number of sequences, we normalized the number of sequences in each sample for the composition and richness analyses and used Fisher’s α-diversity index (Fisher et al., 1943)—a reliable index of species diversity that is independent of sample size (Hubbell, 2001)—for the diversity analyses.
To characterize the stability of bacterial communities in individual rodents, we used the Jaccard index (Jaccard, 1912; see Supplementary Methods). For each individual rodent, we then compared the ‘intraindividual stability’ and the ‘interindividual stability’. We used a generalized estimating equation model to evaluate the effect of the source of variation (within versus between host individuals), sample type and their interaction (independent variable) on the Jaccard index (dependent variable) and compared each model with an intercept-only model by using the model selection approach.
Results
Blood and flea microbiomes
Details on the gerbil and flea samples are given in the Supplementary Results. Despite capturing 511 individuals, only 8 of the juvenile gerbils were recaptured during the summer, possibly because of a high dispersal rate. The minimal number of fleas per host individual was four. Accordingly, we balanced the numbers of juvenile and adult hosts and of flea and blood samples: for molecular analyses, we used 16 individuals (4 from each sex–age combination) and collected, from each individual and at each sampling period (spring/summer), one blood sample, two female fleas and two male fleas. The DNA from the two samples of the same flea sex were pooled. This sampling procedure yielded in total 32 DNA extracts from blood and 64 from fleas. DNA extracts from 59 of these flea samples and from all blood samples were successfully subjected to pyrosequencing, yielding 138–1139, 213–1173 and 155–1167 sequences/sample in the blood, female flea and male flea samples, respectively. The cleaned sequences had lengths ranging from 400 to 494, with a mean of 449 and a s.d. of 7.2. Altogether, 40 bacterial phylotypes were significantly more abundant in the field samples than in the negative controls and represented at least 2% of the total number of sequences detected in any sample. These phylotypes represented 22 genera (Table 2), but only 8 of these were represented by sequences that comprised >20% of the total sequences in a given sample. Bartonella, Wolbachia and Mycoplasma were the most prevalent of these genera, that is, were common in more than one DNA sample and outnumbered other genera in the sample by a factor of ≥10 (Supplementary Figure S1).
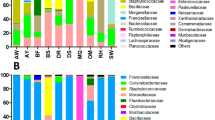
The composition of detected phylotypes is detailed in Table 2, where we distinguish between phylotypes found in only one sample, rare phylotypes and phylotypes that appear in at least 5% of the samples. The latter group, of the most prevalent phylotypes, consisted of only nine phylotypes, of which all but two (namely, Wolbachia spp.) were shared by fleas and host blood samples, and represented five genera. Of these genera, Bartonella and hemotropic Mycoplasma, which are known vector-borne bacteria (Willi et al., 2005; Morick et al., 2010, 2011), were the most common (Figure 1 and Supplementary Figure S1). Methylobacterium and Streptococcus have been previously detected in arthropods and in vertebrates, and may thus be vector-borne bacteria as well. Diaphorobacter was previously found in arthropods but not in vertebrate blood; it is not known as a member of the skin or eye microbiome (Cullen, 2003; Coster et al., 2008; Montiani-Ferreira et al., 2008; Scharschmidt et al., 2009; Grice and Segre, 2011; Hoffmann et al., 2014; Meekins et al., 2014; Tomic-Canic et al., 2014), and therefore its occurrence in the gerbil blood is probably not a result of contamination and deserves further investigation. Seven additional phylotypes were detected in 2–4 samples, representing five genera. Of these seven phylotypes, two shared phylotypes belonged to the genera Staphylococcus and Bradyrhizobium, which were detected previously in rodents. Staphylococcus was also detected in fleas and Bradyrhizobium was also detected in soil and, therefore, could have been acquired by fleas from the soil (for example, via food consumption during the free-living life stages of the fleas). One phylotype was unique to fleas and represents Wolbachia, a genus with known mutual associations with arthropods. Finally, four phylotypes were unique to blood, representing three taxonomic groups (Bradyrhizobium, Halomonas and a member of the Sphingobacteria class) with representative isolates from blood or from other tissues of vertebrate species.

Mean relative abundance of bacterial genera (that is, sequences classified as a certain genus, as percentage of the total number of sequences) detected in the different sample types (female fleas (FF), male fleas (MF) or rodent blood (B)) collected from 16 female and male G. andersoni hosts. The hosts were trapped either as juveniles (JH) or as adults (AH) during the spring (SP), and resampled as adults at the end of summer (SU). The most abundant genera (that is, those comprising at least 20% of the total sequences in at least one DNA sample) are specified, whereas all remaining sequences are represented as ‘others’. Collective data are shown (a) from all host individuals and (b) for only blood samples. *Only class is provided because the phylotype was not classified to lower taxonomic levels.
Determinants of the bacterial community structure
The ‘sample type’ and the ‘sample type’ × ‘sampling season’ interaction were important predictors of the composition of the bacterial community (pseudo-F >6, P<0.0005 for each variable; Figures 1a and 2), and together explained 33% of the variability in phylotype composition among bacterial communities. A SIMPER analysis revealed that two Wolbachia phylotypes (which were detected only in female fleas; Figure 1a and Supplementary Figure S1) and one Bartonella phylotype (which was more abundant in male fleas and in host blood samples than in female fleas; Figure 3) were primarily responsible for the differences between bacterial communities in fleas and in the blood of their hosts. The higher relative abundance of Wolbachia species and the lower relative abundance of Bartonella species in female fleas, as compared with that in male fleas, may result from differences in the number of Bartonella sequences, in the number of Wolbachia sequences or both. However, the mean absolute number of Bartonella sequences found in Wolbachia-free (that is, male) fleas was significantly higher than in Wolbachia-infected (that is, female) fleas (574±49 and 247±45, respectively; Wald χ2=16, P<0.0001).

Nonmetric multidimensional scaling (MDS) ordination of bacterial communities in female fleas (FF; pink), male fleas (MF; blue) and host blood (B; red). Samples were collected from G. andersoni rodents during the spring (SP; empty circles) or summer (SU; filled circles). MDS is based on Bray–Curtis similarities in fourth root transformed abundance data of the different bacterial phylotypes. Each point represents a bacterial community from each sample, and the proximity between points represents the extent of similarity in bacterial community compositions. One outlier, namely, a bacterial community in a male flea, was omitted.

Mean abundance of Bartonella sp., which were the most abundant and influential bacterial phylotype found, in female fleas (FF; pink), male fleas (MF; blue) and rodent blood (B; red). Samples were collected from G. andersoni hosts, trapped during the spring (SP) and summer (SU). Data from all hosts are combined.
The significance of the ‘sample type’ × ‘sampling season’ interaction reflects a seasonal variability in the composition of bacterial communities in female fleas (pseudo-F=4, P<0.01; Figure 2) and in the blood of their hosts (pseudo-F=3, P<0.05; Figures 1b and 2), but not in the male fleas (pseudo-F=0.4, P=0.9; Figure 2). A SIMPER analysis revealed that one Wolbachia phylotype and one Bartonella phylotype, which were both more abundant in the spring than in the summer, were primarily responsible for this variability in female fleas. In the host blood, one Mycoplasma phylotype and one Bartonella phylotype explained 39% of the seasonal variability, with the abundance of Mycoplasma increasing and that of Bartonella decreasing from spring to summer (Figure 1b). However, the ratio between the average and the s.d. of dissimilarity of these two phylotypes was smaller than one, indicating that their contribution to the seasonal variability between bacterial communities in the blood is not highly consistent. After removing the two Wolbachia and one Bartonella phylotypes that were mainly responsible for the observed patterns in the composition of the bacterial communities, none of the independent variables significantly affected the composition of the bacterial phylotypes. The identity of the individual host, its gender, age or the second or third interaction terms between these variables did not have a significant effect on the composition of the bacterial phylotypes, either when all bacterial phylotypes were included or when the most common phylotypes were removed (Figures 1 and 3, Supplementary Figure S1).
The ‘sample type’ was the best predictor of bacterial community diversity (Table 1), and this was higher in female fleas than in male fleas or host blood (Figure 4a). However, after removing the two Wolbachia phylotypes and the one Bartonella phylotype that were mainly responsible for the observed patterns in the community composition, the ‘sample type’ alone was a poor predictor of community diversity (Table 1). Rather, when ignoring the two common phylotypes, the ‘sample type’ × ‘sampling season’ interaction was the best predictor of community diversity, decreasing in female fleas and increasing in host blood between spring and summer (Table 1 and Figure 4b). In explaining the observed variation in the richness of the bacterial phylotypes, the weight of the intercept-only model was similar (ΔQICC <2) to that of the model that included ‘sample type’, and was higher than that of all other models (ΔQICC>2 for the comparison between the intercept-only model and each of the other models). This result suggests that all the tested variables are weakly associated with phylotype richness (Table 1).

Bacterial phylotype diversity (mean Fisher’s α-index) measured for bacterial communities obtained from female fleas, male fleas and host blood samples (FF, MF and B, respectively). Samples were obtained from female (FH) and male (MH) G. andersoni rodent hosts, trapped either as juveniles (JH) or as adults (AH) during the spring (SP), and resampled as adults during the summer (SU). Values below and above the median are indicated by a white and black color, respectively, and values that are more than twice the median are indicated by a gray color. (a) Data of all bacterial communities; (b) data of the bacterial communities excluding the most dominant and influential phylotypes: one Bartonella and two Wolbachia phylotypes.
Stability of bacterial communities in individual hosts
The weights of the intercept-only models were higher (ΔQICC>2) than those of the models that included the ‘source of variation’ or the ‘sample type’ factors, possibly indicating a high variance in the Jaccard index calculated for different combinations of paired communities. Notably, however, the stability of bacterial communities was higher between host individuals than within host individuals (Figure 5).

Comparison of microbiota stability (means±s.e. of the Jaccard index) within and between host individuals.
Discussion
Microbial symbionts play many important roles in the evolution, development, physiology and behavior of animals (McFall-Ngai et al., 2013). However, as most studies of host-associated symbionts have focused on microbial communities in the mammalian gut (Peterson et al., 2009; McFall-Ngai et al., 2013), basic ecological principles shaping these communities remain unknown for the vast majority of species. The characterization of microbial communities associated with arthropod vector-borne bacteria is of particular interest, because these bacteria are members of two distinct communities that may be influenced, simultaneously, by the characteristics of the vector, the characteristics of its host and the temporal variations to which both the vector and the host may be exposed (Hawlena et al., 2013). We report that bacterial communities in fleas and in the blood of their rodent hosts are dominated by flea-borne bacteria and are thus remarkably similar in terms of community structure and stability. Furthermore, although bacterial communities in both the fleas and the rodent hosts were similar—regardless of being isolated from different rodent individuals—they varied seasonally. In fact, possibly because of the presence of Wolbachia only in female fleas, the bacterial communities in the male fleas and in the blood of their rodent hosts were more similar to each other than those in female and male fleas. Taken together, our results suggest that the overall abundance of a certain flea-borne microbe is more likely to be determined by the abundance of endosymbiotic bacteria in the flea, abundance of other flea-borne microbes co-occurring in the flea and in the rodent blood or seasonal changes rather than by rodent characteristics.
Bacterial communities in fleas are remarkably similar to those in the blood of their hosts
Some bacteria in the blood of rodents can potentially be irrelevant to fleas, for example, because they are transmitted via helminths, ticks or other arthropods (Hawlena et al., 2006 and Hawlena, Veciana, and Ribas, unpublished data) or via direct transmission between hosts. Hence, we hypothesized that the bacterial communities in the flea vectors would be substantially different from those in their vertebrate hosts. In addition, because fleas acquire bacteria from the soil (which is characterized by diverse bacterial communities) and because the blood of the rodent hosts is a relatively sterile environment, we hypothesized that bacterial communities in the fleas would show greater species richness and diversity than those in the blood of their hosts.
Our hypotheses were only partly confirmed: although the bacterial communities in the fleas were different from those in the blood of their hosts, the two communities shared seven of the most common phylotypes and both exhibited low and remarkably similar phylotype richness and diversity. These similarities suggest that flea-borne bacteria form the core microbiota in both communities. Regarding Bartonella, experimental transmission confirms that the main Bartonella bacterium in our study is, indeed, flea-borne (Morick et al., 2010, 2011, 2013; Gutiérrez et al., 2014). In addition, the two other common Bartonella phylotypes were detected in both blood and flea samples (Table 2). Finally, we recently reported a pyrosequencing analysis, conducted on the same field samples used here, that targeted the citrate synthase gene to investigate Bartonella phylotype diversity (Gutiérrez et al., 2014). The higher resolution and more specific target gene explored in that study provided us with a greater coverage, including 331 Bartonella phylotypes. Nevertheless, all 14 phylotypes that represented 95% of all reads were observed in both fleas and blood samples (Gutiérrez et al., 2014). With respect to the detected Mycoplasma phylotype, we already demonstrated rodent-to-flea transmission and we are currently conducting a complement experiment to confirm flea-to rodent transmission (C Cohen et al., unpublished data). Similar experiments should be conducted to confirm that the three other shared phylotypes are also flea-borne. Evidence for interspecific interactions among vector-borne microbes have been reported in fleas and ticks and in various vertebrate hosts (Cox, 2001; Clay et al., 2006; Steiner et al., 2008; Jones et al., 2010; Telfer et al., 2010; Gutiérrez et al., 2014; Kedem et al., 2014). It is thus likely that the overall abundance of a specific vector-borne bacterium (for example, Bartonella sp. in our study) is influenced by the abundance of other phylotypes of vector-borne bacteria co-occurring in the vectors and in the blood of their hosts (for example, Mycoplasma sp. in our study).
The Gutiérrez et al. (2014) study was the first and only study, to the best of our knowledge, to have compared bacterial communities in vertebrate blood and in arthropod vectors. That study revealed some differences in Bartonella phylotype composition between flea vectors and blood samples from their rodent hosts. However, the intersexual distinction (that is between female and male fleas) in bacterial communities was similar or even more pronounced than the distinction between bacterial communities in fleas and blood in both macropatterns (that is, different genera, in our study) and micropatterns (that is, different species and genotypes in the study by Gutiérrez et al., 2014) of the community structure. This fact suggests that the bacterial communities in the flea vectors and in the blood of their vertebrate hosts are shaped by similar factors. One possibility is that the blood meal remnants in the gut of fleas are sufficiently close to the blood in live gerbils to present similar conditions (for example, similar substrate, thermal conditions or resources). Accordingly, the body of the rodent or flea may not ‘filter out’ the dominant phylotypes that are introduced by the flea or the rodent, respectively. However, even if bacteria in fleas and in rodents are exposed to different conditions, they may still be exposed to similar selective forces, reducing the possible number of coexisting bacteria via, for example, interspecific competition between bacteria (see, for example, Macaluso et al., 2002; Hawlena et al., 2012; Zouache et al., 2012) or the immunity of the rodent hosts (see, for example, Cox, 2001) and flea vectors (Vallet-Gely et al., 2008) to their bacterial inhabitants. By extrapolation, low bacterial diversity likely characterizes any microbial community in arthropod vectors and in the blood of their vertebrate hosts. This observation may simplify the efforts to control emerged vector-borne pathogens by manipulating the density of their bacterial inhabitants (see, for example, Cirimotich et al., 2011). This is because, in poor bacterial communities, we have only a few candidate species that could affect pathogen densities, and this should ease the screening and experimental procedures.
The composition of bacterial communities in fleas and in the blood of their rodent hosts has only a weak association with host characteristics and exhibits variations from spring to summer
One of the main goals in the study of microbiomes is to investigate the causes for the temporal and spatial variability in the composition of microbial communities. Although some degree of interhost variability was detected (Supplementary Figure S1), we found indications that the variability in bacterial community composition was lower between host individuals than within host individuals, and that it was similar between juvenile and adult hosts and between female and male hosts. From spring to summer, the relative abundance of Wolbachia and Bartonella phylotypes in female fleas decreased, whereas that of Mycoplasma sp. in the blood of the hosts increased, in a manner that was independent of the host identity, their sex or their maturation timeline (Table 1, Figures 1b and 3 and Supplementary Figure S1). Seasonal interhost variability in physiology, immunity and behavioral responses to symbionts is often most pronounced when comparing juveniles with adults and males with females (Hudson and Dobson, 1997; Wilson et al., 2002; Mooring et al., 2004; Hawlena et al., 2005; Altizer et al., 2006; Grassly and Fraser, 2006; Krasnov et al., 2006). However, the fact that bacterial communities in all gerbil groups have passed through similar seasonal changes suggest that these changes may not be linked to changes in the internal environment of the host. Other studies reported that microbial communities show high variability between host individuals, age groups and sexes and/or a relatively high temporal stability (Moran et al., 2005; Costello et al., 2009; Godoy-Vitorino et al., 2010; Oh et al., 2012; Yatsunenko et al., 2012; Faith et al., 2013; van Dongen et al., 2013; Kueneman et al., 2014; Leclaire et al., 2014). The lack of interindividual variations in the composition of bacterial communities in our study may result from the transmission of fleas between hosts via direct interactions, replacement of burrows or free dispersal (Krasnov, 2008). These processes may equilibrate the microbial communities across host individuals, given that vector-borne phylotypes are the core of these communities.
The spring-to-summer variations in bacterial communities in the female fleas may be explained, in part, by the nature of ectoparasitic vectors that are affected by the external environment of their host (Krasnov, 2008). Fleas spend time in the burrows of their rodent hosts during both their free-living (when they consume organic matter) and parasitic (adult) life stages. Thus, the bacteria associated with fleas can be affected by extrinsic factors that vary across seasons. For example, it is possible that the composition of microbial communities changes in response to an increase in flea burden over summer months (Lehmann, 1989) or that, similar to other flea species (Krasnov et al., 2002), S. cleopatrae fleas in the field undergo physiological changes that affect the relative abundance of Wolbachia species in females. The spring-to-summer changes that we found in bacterial communities in the host blood (Figure 1b) may have resulted from accumulation of some bacterial phylotypes—such as the Mycoplasma sp. found in both flea and blood samples—that, once acquired, persist in the host. Indeed, our lab experiments show that Mycoplasma sp. can persist in the blood of an individual G. andersoni rodent for >3 years (C Cohen et al., unpublished data). If resources for these bacterial phylotypes are limited in the blood of the rodent host, then the accumulation of these phylotypes may come at the expense of other phylotypes. To determine whether the observed spring-to-summer variations in flea-borne bacteria reflect a more general temporal pattern, one should sample microbes in additional timeframes and in multiple years and vector species. Reports of climatic influences on microbial abundance and composition in flies, mosquitoes and ticks support a general role of seasonality (Kirstein et al., 1997; Roper et al., 1998; Sutherst, 2004; Wielinga et al., 2006; Ostfeld, 2009; Lalzar et al., 2012). If seasonal variability in microbial community structure indeed plays an important role in determining the overall abundance of vector-borne bacteria, then future investigations of the specific causes of seasonal variability are encouraged.
Wolbachia phylotypes play an important role in shaping the bacterial community in female fleas
The differences in bacterial composition between the sample types and the significantly higher bacterial diversity in female than in male fleas or in the host blood were attributed to the Wolbachia phylotypes. Wolbachia species are common endosymbiotic bacteria in arthropods, exerting several phenotypic effects on their hosts and generally behaving as reproductive parasites that distort the sex ratio of arthropods toward females. Wolbachia bacteria have been previously detected in fleas, but the nature of this association is not yet known (Gorham et al., 2003). In our S. cleopatrae flea samples, Wolbachia phylotypes were found exclusively in females and comprised a significant portion of the bacterial community in all samples collected from female fleas. This finding suggests that Wolbachia are transmitted vertically between fleas and that feminization, male killing or both mechanisms are involved in the Wolbachia–flea relationships (Werren et al., 2008). However, despite the high prevalence of Wolbachia in the female flea population, the flea sex ratio was not distorted toward females (see Supplementary Results). Regardless of the mechanism through which Wolbachia affects flea populations, our results suggest that the presence of Wolbachia affects the load of Bartonella species—which are the most common flea-borne bacteria in our study system. In our study system, the effects of the flea sex and of Wolbachia presence or absence are confounded, as these bacteria were restricted to female fleas. Thus, to confirm a direct negative association between Wolbachia and Bartonella, one should manipulate the abundance of Wolbachia in female fleas and quantify (for example, by using a quantitative PCR analysis) the resulting number of Bartonella cells.
Wolbachia bacteria thus appear to play an important role in shaping natural communities that are associated with flea-borne bacteria. Other vector endosymbionts are known to shape microbial communities in mosquitoes, fleas, aphids and ticks (Philip et al., 1976; Noda et al., 1997; Aksoy, 2000; Macaluso et al., 2002; Gonzalez-Ceron et al., 2003; Scarborough et al., 2005; Cirimotich et al., 2011), indicating, more generally, that the abundance of bacterial endosymbionts are good predictors of the abundance of vector-borne bacteria. From an applied perspective, the sex-related differences in bacterial community structure indicate that male but not female fleas are probably the main source of flea-borne pathogens. The same conclusion may hold true for any arthropod vector that harbors a reproductive parasite, suggesting that efforts to control these vectors should focus on the males.
Summary and conclusion
We used deep sequencing to provide a comprehensive catalog of bacteria that naturally inhabit rodent blood and fleas. We report that, in both the rodent blood and the fleas, bacterial communities within an individual are characterized by a very low diversity and are dominated by flea-borne phylotypes. In addition, the structure of bacterial communities is remarkably similar in fleas and in the blood of their rodent hosts, with the exception of Wolbachia bacteria that are present exclusively in female fleas. Finally, we show that, whereas the structure of communities in both the rodent blood and the fleas is largely independent of host characteristics, both these communities are prone to seasonal variations. These findings are consistent with data on other vector-borne microbes (Philip et al., 1976; Pumpuni et al., 1993; Kirstein et al., 1997; Noda et al., 1997; Roper et al., 1998; Aksoy, 2000; Cox, 2001; Macaluso et al., 2002; Gonzalez-Ceron et al., 2003; Sutherst, 2004; Scarborough et al., 2005; Clay et al., 2006; Steiner et al., 2006; Wielinga et al., 2006; Ostfeld, 2009; Jones et al., 2010; Telfer et al., 2010; Cirimotich et al., 2011; Lalzar et al., 2012). Collectively, they highlight the following candidate factors as affecting the overall abundance of a specific vector-borne microbe: (1) the abundance of microbial endosymbionts of vectors, (2) the abundance of other vector-borne microbes that co-occur in the vector and in the blood of its host and (3) seasonal variation in the host environment. Our findings thus indicate that efforts to prevent the spread of such vector-borne pathogens should focus on monitoring and controlling the three factors specified above.
Accession codes
References
Abramsky Z, Brand S, Rosenzweig ML . (1985). Geographical ecology of gerbilline rodents in sand dune habitats of Israel. J Biogeogr 12: 363–372.
Aksoy S . (2000). Tsetse, a haven for microorganisms. Parasitol Today 16: 114–118.
Alexy KJ, Gassett JW, Osborn DA, Miller KV, Russell SM . (2003). Bacterial fauna of the tarsal tufts of white-tailed deer (Odocoileus virginianus). Am Midl Nat 149: 237–240.
Altizer S, Dobson A, Hosseini P, Hudson P, Pascual M, Rohani P . (2006). Seasonality and the dynamics of infectious diseases. Ecol Lett 9: 467–484.
Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402.
Andreotti R, de Leon AAP, Dowd SE, Guerrero FD, Bendele KG, Scoles GA . (2011). Assessment of bacterial diversity in the cattle tick Rhipicephalus (Boophilus) microplus through tag-encoded pyrosequencing. BMC Microbiol 11: 6.
Azad AF, Beard CB . (1998). Rickettsial pathogens and their arthropod vectors. Emerg Infect Dis 4: 179–186.
Bar Y, Abramsky Z, Gutterman Y . (1981). Diet of gerbilline rodents in the Israeli Desert. J Arid Environ 7: 371–376.
Beachey EH . (1980) Bacterial Adherence. Kluwer Academic Publishers: Dordrecht, The Netherlands.
Bottomley PJ, Cheng HH, Strain SR . (1994). Genetic structure and symbiotic characteristics of a Bradyrhizobium population recovered from a pasture soil. Appl Environ Microbiol 60: 1754–1761.
Burnham KP, Anderson DR . (2002) Model Selection and Multimodel Inference. A Practical Information-Theoretic Approach 2 edn. Springer Science and Business Media: New York.
Chomel BB, Boulouis HJ, Breitschwerdt EB, Kasten RW, Vayssier-Taussat M, Birtles RJ et al. (2009). Ecological fitness and strategies of adaptation of Bartonella species to their hosts and vectors. Vet Res 40: 29.
Cirimotich CM, Dong Y, Clayton AM, Sandiford SL, Souza-Neto JA, Mulenga M et al. (2011). Natural microbe-mediated refractoriness to Plasmodium infection in Anopheles gambiae. Science 332: 855–858.
Clarke KR . (1993). Nonparametric multivariate analyses of changes in community structure. Aust J Ecol 18: 117–143.
Clay K, Fuqua C, Lively CM, Wade MJ . (2006). Microbial community ecology of tick-borne human pathogens. In: Collinge SK, Ray C, (eds) Disease Ecology: Community Structure and Pathogen Dynamics. Oxford University Press: Oxford, pp 41–57.
Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R . (2009). Bacterial community variation in human body habitats across space and time. Science 326: 1694–1697.
Coster ME, Stiles J, Krohne SG, Raskin RE . (2008). Results of diagnostic ophthalmic testing in healthy guinea pigs. J Am Vet Med Assoc 232: 1825–1833.
Cox FEG . (2001). Concomitant infections, parasites and immune responses. Parasitology 122: S23–S38.
Cullen CL . (2003). Normal ocular features, conjunctival microflora and intraocular pressure in the Canadian beaver (Castor canadensis). Vet Ophthalmol 6: 279–284.
de la Fuente J, Blouin EF, Kocan KM . (2003). Infection exclusion of the rickettsial pathogen Anaplasma marginale in the tick vector Dermacentor variabilis. Clin Diagn Lab Immunol 10: 182–184.
Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL et al. (2013). The long-term stability of the human gut microbiota. Science 341: 44-+.
Fierer N, Hamady M, Lauber CL, Knight R . (2008). The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci USA 105: 17994–17999.
Fisher RA, Cobet AS, Williams CB . (1943). The relation between the number of species and the number of individals in a random sample of an animal population. J Anim Ecol 12: 42–58.
Gavish Y, Kedem H, Messika I, Cohen C, Toh E, Munro D et al. (2014). Association of host and microbial species diversity across spatial scales in desert rodent communities. PLoS One 9: e109677.
Gill JJ, Sabour PM, Gong JH, Yu H, Leslie KE, Griffiths MW . (2006). Characterization of bacterial populations recovered from the teat canals of lactating dairy and beef cattle by 16S rRNA gene sequence analysis. FEMS Microbiol Ecol 56: 471–481.
Godoy-Vitorino F, Goldfarb KC, Brodie EL, Garcia-Amado MA, Michelangeli F, Dominguez-Bello MG . (2010). Developmental microbial ecology of the crop of the folivorous hoatzin. ISME J 4: 611–620.
Gonzalez-Ceron L, Santillan F, Rodriguez MH, Mendez D, Hernandez-Avila JE . (2003). Bacteria in midguts of field-collected Anopheles albimanus block Plasmodium vivax sporogonic development. J Med Entomol 40: 371–374.
Gorham CH, Fang QQ, Durden LA . (2003). Wolbachia endosymbionts in fleas (Siphonaptera). J Parasitol 89: 283–289.
Grassly NC, Fraser C . (2006). Seasonal infectious disease epidemiology. Proc Biol Sci 273: 2541–2550.
Grice EA, Segre JA . (2011). The skin microbiome. Nat Rev Microbiol 9: 244–253.
Gutiérrez R, Moric D, Cohen C, Hawlena H, Harrus S . (2014). The effect of ecological and temporal factors on the composition of Bartonella infection in rodents and their fleas. ISME J 8: 1598–1608.
Gutiérrez R, Morick D, Cohen C, Hawlena H, Harrus S . (2014). The effect of ecological and temporal factors on the composition of Bartonella infection in rodents and their fleas. ISME J 8: 1598–1608.
Hawlena H, Abramsky Z, Krasnov BR . (2005). Age-biased parasitism and density-dependent distribution of fleas (Siphonaptera) on a desert rodent. Oecologia 146: 200–208.
Hawlena H, Abramsky Z, Krasnov BR . (2006). Ectoparasites and age-dependent survival in a desert rodent. Oecologia 148: 30–39.
Hawlena H, Bashey F, Lively CM . (2012). Bacteriocin-mediated interactions within and between coexisting species. Ecol Evol 2: 2516–2521.
Hawlena H, Rynkiewicz E, Toh E, Alfred A, Durden LA, Hastriter MW et al. (2013). The arthropod, but not the vertebrate host or its environment, dictates bacterial community composition of fleas and ticks. ISME J 7: 221–223.
Heise SR, Elshahed MS, Little SE . (2010). Bacterial diversity in Amblyomma americanum (Acari: Ixodidae) with a focus on members of the genus Rickettsia. J Med Entomol 47: 258–268.
Hoffmann AR, Patterson AP, Diesel A, Lawhon SD, Ly HJ, Stephenson CE et al. (2014). The skin microbiome in healthy and allergic dogs. PloS One 9: e83197.
Hornok S, Foldvari G, Elek V, Naranjo V, Farkas R, de la Fuente J . (2008). Molecular identification of Anaplasma marginale and rickettsial endosymbionts in blood-sucking flies (Diptera : Tabanidae, Muscidae) and hard ticks (Acari : Ixodidae). Vet Parasitol 154: 354–359.
Hubbell SP . (2001) The Unified Neutral Theory of Biodiversity and Biogeography. Princeton University Press: Princeton, NJ.
Hudson PJ, Dobson AP . (1997). Host-parasite processes and demographic consequences. In: Clayton DH, Moore J, (eds) Host-Parasite Evolution. General Principles and Avian Models. Oxford University Press: New York, pp 128–154.
Jaccard P . (1912). The distribution of the flora in the alpine zone. New Phytol 11: 37–50.
Jones RT, Knight R, Martin AP . (2010). Bacterial communities of disease vectors sampled across time, space, and species. ISME J 4: 223–231.
Kedem H, Cohen C, Messika I, Einav M, Pilosof S, Hawlena H . (2014). Multiple effects of host species diversity on co-existing host-specific and host-opportunistic microbes. Ecology 95: 1173–1183.
Kirstein F, Rijpkema S, Molkenboer M, Gray JS . (1997). Local variations in the distribution and prevalence of Borrelia burgdorferi sensu lato genomospecies in Ixodes ricinus ticks. Appl Environ Microbiol 63: 1102–1106.
Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R et al. (2011). Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci USA 108: 4578–4585.
Konig H . (2006). Bacillus species in the intestine of termites and other soil invertebrates. J Appl Microbiol 101: 620–627.
Krasnov BR, Burdelova NV, Shenbrot GI, Khokhlova IS . (2002). Annual cycles of four flea species in the central Negev desert. Med Vet Entomol 16: 266–276.
Krasnov BR, Stanko M, Morand S . (2006). Age-dependent flea (Siphonaptera) parasitism in rodents: a host's life history matters. J Parasitol 92: 242–248.
Krasnov BR . (2008) Functional and Evolutionary Ecology of Fleas: A Model for Ecological Parasitology. Cambridge University Press: Cambridge.
Kueneman JG, Parfrey LW, Woodhams DC, Archer HM, Knight R, McKenzie VJ . (2014). The amphibian skin-associated microbiome across species, space and life history stages. Mol Ecol 23: 1238–1250.
Labeda DP, Testa RT, Lechevalier MP, Lechevalier HA . (1984). Saccharothrix: a new genus of the Actinomycetales related to Nocardiopsis. Int J Syst Bacteriol 34: 426–431.
Lalzar I, Harrus S, Mumcuoglu KY, Gottlieb Y . (2012). Composition and seasonal variation of Rhipicephalus turanicus and Rhipicephalus sanguineus bacterial communities. Appl Environ Microbiol 78: 4110–4116.
Leclaire S, Nielsen JF, Drea CM . (2014). Bacterial communities in meerkat anal scent secretions vary with host sex, age, and group membership. Behav Ecol doi:10.1093/beheco/aru109.
Lehmann T . (1989) Population Biology of the Flea Synosternus cleopatra with Emphasis on Host-Parasite Relations (in Hebrew, with English summary). The Hebrew University of Jerusalem: Israel, Jerusalem.
Lemon SM, Sparling PF, Hamburg MA, Relman DA, Choffne ER, Mack A . (2008) Vector-Borne Diseases: Understanding the Environmental, Human Health, and Ecological Connections, Workshop Summary 1st edn vol. 1. National Academies Press: Washington, DC.
Lipsitch M, Siller S, Nowak MA . (1996). The evolution of virulence in pathogens with vertical and horizontal transmission. Evolution 50: 1729–1741.
Lively CM, Clay K, Wade MJ, Fuqua C . (2005). Competitive co-existence of vertically and horizontally transmitted parasites. Evol Ecol Res 7: 1183–1190.
Macaluso KR, Sonenshine DE, Ceraul SM, Azad AF . (2002). Rickettsial infection in Dermacentor variabilis (Acari : Ixodidae) inhibits transovarial transmission of a second Rickettsia. J Med Entomol 39: 809–813.
Magurran AE . (1988) Ecological Diversity and Its Measurement. Princeton University Press: Princeton, NJ.
Maidak BL, Olsen GJ, Larsen N, Overbeek R, McCaughey MJ, Woese CR . (1996). The Ribosomal Database Project (RDP). Nucleic Acids Res 24: 82–85.
Martin MO, Gilman FR, Weiss SL . (2010). Sex-specific asymmetry within the cloacal microbiota of the striped plateau lizard, Sceloporus virgatus. Symbiosis 51: 97–105.
McFall-Ngai M, Hadfield MG, Bosch TCG, Carey HV, Domazet-Loso T, Douglas AE et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc Natl Acad Sci USA 110: 3229–3236.
Meekins JM, Eshar D, Rankin AJ . (2014). Tear production, intraocular pressure, and conjunctival bacterial flora in a group of captive black-tailed prairie dogs (Cynomys ludovicianus). Vet Ophthalmol doi:10.1111/vop.12226.
Montiani-Ferreira F, Truppel J, Tramontin MH, Vilani RGD, Lange RR . (2008). The capybara eye: clincial tests, anatomic and biometric features. Vet Ophthalmol 11: 386–394.
Mooring MS, Blumstein DT, Stoner CJ . (2004). The evolution of parasite-defence grooming in ungulates. Biol J Linn Soc 81: 17–37.
Moquin SA, Garcia JR, Brantley SL, Takacs-Vesbach CD, Shepherd UL . (2012). Bacterial diversity of bryophyte-dominant biological soil crusts and associated mites. J Arid Environ 87: 110–117.
Moran D, Turner SJ, Clements KD . (2005). Ontogenetic development of the gastrointestinal microbiota in the marine herbivorous fish Kyphosus sydneyanus. Microb Ecol 49: 590–597.
Moreno CX, Moy F, Daniels TJ, Godfrey HP, Cabello FC . (2006). Molecular analysis of microbial communities identified in different developmental stages of Ixodes scapularis ticks from Westchester and Dutchess Counties, New York. Environ Microbiol 8: 761–772.
Morick D, Krasnov BR, Khokhlova IS, Shenbrot GI, Kosoy MY, Harrus S . (2010). Bartonella genotypes in fleas (Insecta: Siphonaptera) collected from rodents in the Negev Desert, Israel. Appl Environ Microbiol 76: 6864–6869.
Morick D, Krasnov BR, Khokhlova IS, Gottlieb Y, Harrus S . (2011). Investigation of Bartonella acquisition and transmission in Xenopsylla ramesis fleas (Siphonaptera: Pulicidae). Mol Ecol 20: 2864–2870.
Morick D, Krasnov BR, Khokhlova IS, Gottlieb Y, Harrus S . (2013). Transmission dynamics of Bartonella sp Strain OE 1-1 in Sundevall's Jirds (Meriones crassus). Appl Environ Microbiol 79: 1258–1264.
Moriyama K, Ando C, Tashiro K, Kuhara S, Okamura S, Nakano S et al. (2008). Polymerase chain reaction detection of bacterial 16S rRNA gene in human blood. Microbiol Immunol 52: 375–382.
Muegge BD, Kuczynski J, Knights D, Clemente JC, Gonzalez A, Fontana L et al. (2011). Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332: 970–974.
Netusil J, Zakovska A, Horvath R, Dendis M, Janouskovcova E . (2005). Presence of Borrelia burgdorferi sensu lato in mites parasitizing small rodents. Vector Borne Zoonotic Dis 5: 227–232.
Noda H, Munderloh UG, Kurtti TJ . (1997). Endosymbionts of ticks and their relationship to Wolbachia spp. and tick-borne pathogens of humans and animals. Appl Environ Microbiol 63: 3926–3932.
Oh J, Conlan S, Polley EC, Segre JA, Kong HH . (2012). Shifts in human skin and nares microbiota of healthy children and adults. Genome Med 4: 77.
Olofsson TC, Vasquez A . (2009). Phylogenetic comparison of bacteria isolated from the honey stomachs of honey bees Apis mellifera and bumble bees Bombus spp. J Apic Res 48: 233–237.
Osei-Poku J, Mbogo CM, Palmer WJ, Jiggins FM . (2012). Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya. Mol Ecol 21: 5138–5150.
Ostfeld RS . (2009). Climate change and the distribution and intensity of infectious diseases. Ecology 90: 903–905.
Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA et al. (2009). The NIH human microbiome project. Genome Res 19: 2317–2323.
Philip RN, Casper EA, Ormsbee RA, Peacock MG, Burgdorfer W . (1976). Microimmunofluorescence test for serological study of rocky mountain spotted fever and typhus. J Clin Microbiol 3: 51–61.
Pidiyar VJ, Jangid K, Patole MS, Shouche YS . (2004). Studies on cultured and uncultured microbiota of wild Culex quinquefasciatus mosquito midgut based on 16s ribosomal RNA gene analysis. Am J Trop Med Hyg 70: 597–603.
Pumpuni CB, Beier MS, Nataro JP, Guers LD, Davis JR . (1993). Plasmodium falciparum: inhibition of sporogonic development in Anopheles stephensi by gram-negative bacteria. Exp Parasitol 77: 195–199.
Rani A, Sharma A, Rajagopal R, Adak T, Bhatnagar RK . (2009). Bacterial diversity analysis of larvae and adult midgut microflora using culture-dependent and culture-independent methods in lab-reared and field-collected Anopheles stephensi-an Asian malarial vector. BMC Microbiol 9: 96.
Reed DL, Hafner MS . (2002). Phylogenetic analysis of bacterial communities associated with ectoparasitic chewing lice of pocket gophers: a culture-independent approach. Microb Ecol 44: 78–93.
Reeves WK, Dowling APG, Dasch GA . (2006). Rickettsial agents from parasitic Dermanyssoidea (Acari : Mesostigmata). Exp Appl Acarol 38: 181–188.
Reinhold-Hurek B, Hurek T . (2000). Reassessment of the taxonomic structure of the diazotrophic genus Azoarcus sensu lato and description of three new genera and new species, Azovibrio restrictus gen. nov., sp nov., Azospira oryzae gen. nov., sp nov and Azonexus fungiphilus gen. nov., sp nov. Int J Syst Evol Microbiol 50: 649–659.
Roeselers G, Mittge EK, Stephens WZ, Parichy DM, Cavanaugh CM, Guillemin K et al. (2011). Evidence for a core gut microbiota in the zebrafish. ISME J 5: 1595–1608.
Roper C, Richardson W, Elhassan IM, Giha H, Hviid L, Satti GMH et al. (1998). Seasonal changes in the Plasmodium falciparum population in individuals and their relationship to clinical malaria: a longitudinal study in a Sudanese village. Parasitology 116: 501–510.
Ryan MP, Pembroke JT, Adley CC . (2011). Genotypic and phenotypic diversity of Ralstonia pickettii and Ralstonia insidiosa isolates from clinical and environmental sources including High-purity Water. Diversity in Ralstonia pickettii. BMC Microbiol 11: 194.
Saag P, Tilgar V, Mänd R, Kilgas P, Mägi M . (2011). Plumage bacterial assemblages in a breeding wild passerine: relationships with ecological factors and body condition. Microb Ecol 61: 740–749.
Salminen MK, Rautelin H, Tynkkynen S, Poussa T, Saxelin M, Valtonen V et al. (2004). Lactobacillus bacteremia, clinical significance, and patient outcome, with special focus on Probiotic L-Rhamnosus GG. Clin Infect Dis 38: 62–69.
Scarborough CL, Ferrari J, Godfray HCJ . (2005). Aphid protected from pathogen by endosymbiont. Science 310: 1781–1781.
Scharschmidt TC, List K, Grice EA, Szabo R, Program NCS, Renaud G et al. (2009). Matriptase-deficient mice exhibit ichthyotic skin with a selective shift in skin microbiota. J Invest Dermatol 129: 2435–2442.
Schloss PD, Gevers D, Westcott SL . (2011). Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One 6: e27310.
Sharma P, Sharma S, Maurya RK, De TD, Thomas T, Lata S et al. (2014). Salivary glands harbor more diverse microbial communities than gut in Anopheles culicifacies. Parasit Vectors 7: 235.
Stackebrandt E, Goebel BM . (1994). A place for DNA-DNA reassociation and 16s ribosomal RNA sequence analysis in the present species definition in bacteriology. Int J Syst Bacteriol 44: 846–849.
Steiner FE, Pinger RR, Vann CN, Abley MJ, Sullivan B, Grindle N et al. (2006). Detection of Anaplasma phagocytophilum and Babesia odocoilei DNA in Ixodes scapularis (Acari: Ixodidae) collected in Indiana. J Med Entomol 43: 437–442.
Steiner FE, Pinger RR, Vann CN, Grindle N, Civitello D, Clay K et al. (2008). Infection and co-infection rates of Anaplasma phagocytophilum variants, Babesia spp., Borrelia burgdorferi, and the rickettsial endosymbiont in Ixodes scapularis (Acari: Ixodidae) from sites in Indiana, Maine, Pennsylvania, and Wisconsin. J Med Entomol 45: 289–297.
Stevens DA, Hamilton JR, Johnson N, Kim KK, Lee JS . (2009). Halomonas, a newly recognized human pathogen causing infections and contamination in a dialysis center three new species. Medicine 88: 244–249.
Sutherst RW . (2004). Global change and human vulnerability to vector-borne diseases. Clin Microbiol Rev 17: 136–173.
Telfer S, Lambin X, Birtles R, Beldomenico P, Burthe S, Paterson S et al. (2010). Species interactions in a parasite community drive infection risk in a wildlife population. Science 330: 243–246.
Tomic-Canic M, Perez-Perez GI, Blumenberg M . (2014). Cutaneous microbiome studies in the times of affordable sequencing. J Dermatol Sci 75: 82–87.
Torsvik V, Goksoyr J, Daae FL . (1990). High diversity in DNA of soil bacteria. Appl Environ Microbiol 56: 782–787.
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE et al. (2009). A core gut microbiome in obese and lean twins. Nature 457: 480–U487.
Vallet-Gely I, Lemaitre B, Boccard F . (2008). Bacterial strategies to overcome insect defences. Nat Rev Microbiol 6: 302–313.
van Dongen WFD, White J, Brandl HB, Moodley Y, Merkling T, Leclaire S et al. (2013). Age-related differences in the cloacal microbiota of a wild bird species. BMC Ecol 13: 11.
van Overbeek L, Gassner F, van der Plas CL, Kastelein P, Rocha UND, Takken W . (2008). Diversity of Ixodes ricinus tick-associated bacterial communities from different forests. FEMS Microbiol Ecol 66: 72–84.
Voigt CC, Caspers B, Speck S . (2005). Bats, bacteria, and bat smell: Sex-specific diversity of microbes in a sexually selected scent organ. J Mammal 86: 745–749.
Werren JH, Baldo L, Clark ME . (2008). Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol 6: 741–751.
Wielinga PR, Gaasenbeek C, Fonville M, de Boer A, de Vries A, Dimmers W et al. (2006). Longitudinal analysis of tick densities and Borrelia, Anaplasma, and Ehrlichia infections of Ixodes ricinus ticks in different habitat areas in the Netherlands. Appl Environ Microbiol 72: 7594–7601.
Willi B, Boretti FS, Cattori V, Tasker S, Meli ML, Reusch C et al. (2005). Identification, molecular characterization, and experimental transmission of a new hemoplasma isolate from a cat with hemolytic anemia in Switzerland. J Clin Microbiol 43: 2581–2585.
Wilson K, Bjørnstad ON, Dobson AP, Merler S, Poglayen G, Randolph SE et al. (2002). Heterogeneities in macroparasite infections: patterns and processes. In: Hudson PJ, Rizzoli A, Grenfell BT, Heesterbeek H, Dobson AP, (eds) The Ecology of Wildlife Disease. Oxford University Press: Oxford, pp 6–44.
Woods JE, Brewer MM, Hawley JR, Wisnewski N, Lappin MR . (2005). Evaluation of experimental transmission of 'Candidatus Mycoplasma haemominutum' and Mycoplasma haemofelis by Ctenocephalides felis to cats. Am J Vet Res 66: 1008–1012.
Woods JE, Wisnewski N, Lappin MR . (2006). Attempted transmission of 'Candidatus Mycoplasma haemominutum' and Mycoplasma haemofelis by feeding cats infected Ctenocephalides felis. Am J Vet Res 67: 494–497.
Worthen PL, Gode CJ, Graf J . (2006). Culture-independent characterization of the digestive-tract microbiota of the medicinal leech reveals a tripartite symbiosis. Appl Environ Microbiol 72: 4775–4781.
Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M et al. (2012). Human gut microbiome viewed across age and geography. Nature 486: 222-+.
Yokota A, Tamura T, Hasegawa T, Huang LH . (1993). Catenuloplanes japonicus gen-nov, sp-nov, nom rev, a new genus of the order Actinomycetales. Int J Syst Bacteriol 43: 805–812.
Yuan DT . (2010). A metagenomic study of the tick midgut. MS thesis, University of Texas: Houston.
Zouache K, Michelland RJ, Failloux AB, Grundmann GL, Mavingui P . (2012). Chikungunya virus impacts the diversity of symbiotic bacteria in mosquito vector. Mol Ecol 21: 2297–2309.
Acknowledgements
We thank M Einav, Z Sigal, E Hyams and A Tsairi for valuable advice during this study. This study was supported by Marie Curie Career Integration Grant Number FP7-293713 and by United States-Israel Binational Science Foundation (Grant Number 2012063). This is publication number 856 of the Mitrani Department of Desert Ecology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article

Cite this article
Cohen, C., Toh, E., Munro, D. et al. Similarities and seasonal variations in bacterial communities from the blood of rodents and from their flea vectors. ISME J 9, 1662–1676 (2015). https://doi.org/10.1038/ismej.2014.255
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2014.255
This article is cited by
-
Identification of microbial taxa present in Ctenocephalides felis (cat flea) reveals widespread co-infection and associations with vector phylogeny
Parasites & Vectors (2022)
-
Wolbachia infection dynamics in a natural population of the pear psyllid Cacopsylla pyri (Hemiptera: Psylloidea) across its seasonal generations
Scientific Reports (2022)
-
Ontogeny, species identity, and environment dominate microbiome dynamics in wild populations of kissing bugs (Triatominae)
Microbiome (2020)
-
From endosymbionts to host communities: factors determining the reproductive success of arthropod vectors
Oecologia (2017)
-
Path analyses of cross-sectional and longitudinal data suggest that variability in natural communities of blood-associated parasites is derived from host characteristics and not interspecific interactions
Parasites & Vectors (2015)
