
Abstract
Marine planktonic cyanobacteria capable of fixing molecular nitrogen (termed ‘diazotrophs’) are key in biogeochemical cycling, and the nitrogen fixed is one of the major external sources of nitrogen to the open ocean. Candidatus Atelocyanobacterium thalassa (UCYN-A) is a diazotrophic cyanobacterium known for its widespread geographic distribution in tropical and subtropical oligotrophic oceans, unusually reduced genome and symbiosis with a single-celled prymnesiophyte alga. Recently a novel strain of this organism was also detected in coastal waters sampled from the Scripps Institute of Oceanography pier. We analyzed the metagenome of this UCYN-A2 population by concentrating cells by flow cytometry. Phylogenomic analysis provided strong bootstrap support for the monophyly of UCYN-A (here called UCYN-A1) and UCYN-A2 within the marine Crocosphaera sp. and Cyanothece sp. clade. UCYN-A2 shares 1159 of the 1200 UCYN-A1 protein-coding genes (96.6%) with high synteny, yet the average amino-acid sequence identity between these orthologs is only 86%. UCYN-A2 lacks the same major pathways and proteins that are absent in UCYN-A1, suggesting that both strains can be grouped at the same functional and ecological level. Our results suggest that UCYN-A1 and UCYN-A2 had a common ancestor and diverged after genome reduction. These two variants may reflect adaptation of the host to different niches, which could be coastal and open ocean habitats.
Similar content being viewed by others


Introduction
Marine pelagic cyanobacteria play a major role in biogeochemical cycling of carbon and nitrogen in the ocean. Prochlorococcus and Synechococcus together are the most abundant phototrophic prokaryotes on Earth, and are responsible for a major fraction of oceanic carbon fixation (Partensky et al., 1999; Scanlan and West, 2002; Scanlan, 2003; Johnson et al., 2006). Likewise, cyanobacteria capable of fixing molecular nitrogen (‘diazotrophs’) dominate global oceanic N2 fixation although they are typically orders of magnitude less abundant than Prochlorococcus or Synechococcus (Zehr and Paerl, 2008; Zehr and Kudela, 2011; Voss et al., 2013). Together with upward fluxes of deep-water NO3− to the surface ocean, diazotrophs supply the nitrogen requirement of primary productivity and quantitatively balance losses by sinking of organic material, which can sequester CO2 from the atmosphere to deep waters (Karl et al., 1997; Sohm et al., 2011).
There are several groups of quantitatively significant diazotrophic cyanobacteria in the open ocean, all of which thrive mainly in tropical and subtropical latitudes (Stal, 2009). Traditionally, the filamentous, aggregate-forming cyanobacterium Trichodesmium sp. was viewed as the most important oceanic N2 fixer, based on its wide distribution and direct measurements of its N2 fixation capacity (Dugdale et al., 1961; Capone et al., 1997; Bergman et al., 2013). Other diazotrophic cyanobacteria discovered in early microscopic studies are the filamentous heterocyst-forming types of the Richelia and Calothrix lineages, which live in symbioses with several different diatom species (Villareal, 1992; Janson et al., 1999; Foster and Zehr, 2006). More recently, the application of molecular approaches resulted in the discovery of unexpected and unusual cyanobacteria involved in oceanic N2 fixation (Zehr et al., 1998, 2001). These have usually been grouped as ‘unicellular’ diazotrophic cyanobacteria, but, among them, different types have very different lifestyles, with Crocosphaera watsonii being photosynthetic and mostly free-living cells (but see Foster et al., 2011), whereas UCYN-A (Candidatus Atelocyanobacterium thalassa) is a photoheterotroph that is symbiotic with prymnesiophyte algae (Thompson et al., 2012). While the major biogeochemical role of all diazotrophic cyanobacteria is to provide new nitrogen to the system, their different lifestyles suggest important differences regarding their distribution in the ocean, and the fate of the fixed nitrogen and carbon (Glibert and Bronk, 1994; Scharek et al., 1999; Mulholland, 2007).
As a diazotrophic cyanobacterium, UCYN-A (termed UCYN-A1 from here on) is remarkable in several ways. Although somewhat closely related to Cyanothece sp. strain ATCC 51142, the UCYN-A1 genome is only 1.44 Mb and lacks many genes including whole metabolic pathways and proteins, such as the oxygen-evolving photosystem II and RuBisCO, that is, features that normally define cyanobacteria (Tripp et al., 2010). The recent identification of a symbiotic eukaryotic prymnesiophyte partner, to which UCYN-A1 provides fixed nitrogen while receiving carbon in return, is the first known example of a symbiosis between a cyanobacterium and a prymnesiophyte alga (Thompson et al., 2012). Further, UCYN-A1 can be detected in colder and deeper waters compared with other major N2 fixers like Trichodesmium sp. and C. watsonii (Needoba et al., 2007; Langlois et al., 2008; Rees et al., 2009; Moisander et al., 2010; Diez et al., 2012), and is also abundant in some coastal waters (Mulholland et al., 2012).
There is now evidence that there are at least three nifH lineages of UCYN-A in the ocean (Thompson et al., 2014). These different clades were previously unrecognized because their nifH amino-acid sequences are nearly identical, with sequence variation primarily only occurring in the third base pair of each codon (Thompson et al., 2014). It is unknown whether these strains are different metabolic variants of UCYN-A, analogous to observations in free-living cyanobacteria like Prochlorococcus and Synechococcus, which have extensive heterogeneity in their genome contents that enable them to occupy different niches along gradients of nutrients and light (Moore et al., 1998; Ahlgren et al., 2006; Kettler et al., 2007). Phylotype ‘UCYN-A2’ shares only 95% nifH nucleotide similarity with UCYN-A1, and was discovered to be abundant and actively expressing nifH off the Scripps Institute of Oceanography (SIO) pier. This habitat seems to generally lack UCYN-A1 and has environmental conditions that clearly differ from the tropical/subtropical oligotrophic open ocean during most times of the year (Chavez et al., 2002). UCYN-A2 is associated with a prymnesiophyte host that is closely related to, but not identical to, the UCYN-A1 host (Thompson et al., 2014). Interestingly, the known 18S rRNA gene sequences of the UCYN-A2 host generally fall into a ‘coastal’ cluster whereas the UCYN-A1 host sequences almost exclusively cluster with sequences recovered from open ocean environments (Thompson et al., 2014). Further, both UCYN-A1 and its host appear to be significantly smaller than UCYN-A2 and its host (Thompson et al., 2014). On the basis of these findings, Thompson et al. (2014) suggested that UCYN-A1 could be an oligotrophic open ocean ecotype, whereas UCYN-A2 could possibly be more adapted to coastal waters.
The present study is the first opportunity to characterize the metabolic potential of a new clade of UCYN-A, by analyzing the metagenome of a UCYN-A2 population sampled from waters off the SIO pier. This enabled us to test whether habitat differences, or a distinct symbiont–host relationship, are reflected in genome features that distinguish UCYN-A2 from UCYN-A1, and whether UCYN-A2 has the same lack of genes as UCYN-A1. With the availability of the new UCYN-A2 metagenome, it was also possible to perform phylogenomic analyses (including 135 proteins), to determine whether UCYN-A2 and UCYN-A1 form a monophyletic group, and to establish how these two organisms are related to other cyanobacteria.
Materials and methods
Sampling
After the initial detection of a new nifH phylotype similar to UCYN-A1 in coastal waters off Scripps Pier and its classification as a new strain (UCYN-A2, Thompson et al., 2014), we used the previously described cell-sorting approach (Zehr et al., 2008; Thompson et al., 2012) to obtain cell sorts enriched in UCYN-A2 for genome sequencing. Surface water samples (10 l) were taken at Scripps Pier with a bucket, gently poured into a polypropylene bottle and immediately transferred to the laboratory at Scripps. The sample was then concentrated by gentle vacuum filtration through a 0.22-μm-pore-size polycarbonate filter and cells resuspended by vortexing the filter in 50 ml of sterile-filtered seawater. The concentrate was flash-frozen in liquid nitrogen and shipped to the University of California, Santa Cruz, USA.
Fluorescence-activated cell sorting and nifH quantitative PCR (qPCR) and genome amplification
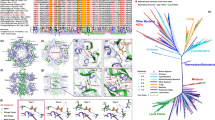
The concentrated seawater samples were thawed at room temperature and briefly vortexed again immediately prior to cell sorting. Seawater samples were pre-filtered using 50-μm-mesh-size CellTrics filters (Partec, Swedesboro, NJ, USA) to prevent clogging of the nozzle (70 μm diameter) with large particles. Samples were analyzed in logarithmic mode with an Influx Cell Sorter (BD Biosciences, San Jose, CA, USA). Flow cytometry sorting gates were defined using forward scatter (a proxy for cell size) and chlorophyll fluorescence at 692 nm (Figure 1). Chlorophyll autofluorescence was excited using a 200-mW, 488-nm sapphire laser (Coherent, Santa Clara, CA, USA).

Work flow diagram describing the cell-sorting, genome-sequencing and assembly approach used in this study. The chosen FCM sort gate was determined in earlier experiments by screening different sorted populations for the presence of UCYN-A2 nifH by qPCR, as described previously. The PRICE assembly was carried out as described in Ruby et al. (2013).
A UCYN-A2-specific qPCR assay (Thompson et al., 2014) was used to screen sorted events within each gate (between 100 and 200 events). Cells were sorted directly into aliquots of 10-μl 5-kDa filtered nuclease-free water, and then amended with qPCR 1 × Universal PCR master mix (Applied Biosystems, Foster City, CA, USA) to a total reaction volume of 25 μl, including UCYN-A2-specific forward and reverse primers (0.4 μM final concentration), as well as TaqMan (Life Technologies Corp., Grand Island, NY, USA) probes (0.2 μM final concentration). qPCR reactions were conducted in a 7500 real-time PCR instrument (Applied Biosystems). Reaction and thermal-cycling conditions were as described previously (Moisander et al., 2010; Thompson et al., 2014). Abundances of nifH gene copies were quantified relative to standard curves comprising amplification of linearized plasmids containing inserts of the target nifH gene, and abundances of gene copies per sample calculated as described by Short and Zehr (2005). Standards were made from serial dilutions of plasmids in nuclease-free water (range: 1–103 nifH gene copies per reaction), with 2 μl of each dilution added up to 25 μl of qPCR (total volume) mixtures. Duplicates of each standard were included with each set of samples run on the qPCR instrument, as well as at least two no-template controls.
Using this approach, we detected a sort region relatively enriched in UCYN-A2 but still containing other organisms besides the target (Figure 1). This region appears to include single UCYN-A2 cells rather than populations in the picoeukaryote-size fraction as described in Thompson et al. (2014) (Figure 1). The disruption of the UCYN-A symbiotic association appears to be a typical result of the concentration and freezing protocol (Thompson et al., 2012), and proved advantageous for our genome amplification and assembly. A sample taken on 31 May 2011 was used to obtain a cell sort enriched in UCYN-A2 for genome amplification (Figure 1). Approximately 3.5 × 104 events were sorted into a 1.5-ml microcentrifuge tube containing 90 μl of TE buffer. Cells were pelleted at 14 000 r.p.m. (21 000 × g) for ∼45 min and the supernatant was discarded. We used a Qiagen REPLI-g Midi kit (Valencia, CA, USA) for cell lysis and amplification of genomic DNA, following the manufacturer’s recommendations with few modifications. Briefly, the pelleted cells were resuspended in 3.5 μl phosphate-buffered saline buffer and 3.5 μl buffer D2 (0.09 M dithiothreitol), incubated at 65 °C for 5 min, and immediately stored on ice after adding the kit-provided ‘stop buffer’. The amplification reaction was carried out in a thermal cycler at 30 °C for 6 h after addition of 40 μl Repli G mastermix to the tube. The quality, size and quantity of the amplified DNA were checked using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and again quantified using Pico Green (Invitrogen Corp., Carlsbad, CA, USA). The suitability of this sample for a genome-sequencing run was indicated by the presence of 106 nifH gene copies of UCYN-A2 per μl, measured by qPCR.
Illumina sequencing
Library preparation and paired-end sequencing were performed at the BioMicro Center of the Massachusetts Institute of Technology (MIT, http://openwetware.org/wiki/BioMicroCenter:Sequencing). The DNA sample was split into two equal aliquots and prepared for sequencing using the SPRI works system (Beckman Coulter Genomics, Danvers, MA, USA) with 150–350 and 250–550 bp inserts. Ligated libraries were amplified and molecular barcodes added. Samples were pooled and sequenced on an Illumina (San Diego, CA, USA) MiSeq v1 flowcell with 151 bp of sequence read in each direction. Fastq files (Illumina v1.5) were prepared and separated into the individual libraries, allowing one mismatch with the barcode sequences. Post-run quality control includes confirmation of low sequencing error rates by analyzing phiX spike sequences, checking for significant contamination from human, mouse, yeast and Escherichia coli, and confirming the presence of only the expected barcodes.
Please see the Supplementary Material section for a detailed description of sequence assembly, annotation and phylogenomic analyses. This sequencing project has been deposited at DDBJ/EMBL/GenBank under the organism name ‘Candidatus Atelocyanobacterium thalassa isolate SIO64986’, accession number JPSP01000000.
Results
The aligned UCYN-A2 scaffolds to the UCYN-A1 reference chromosome covered nearly the entire UCYN-A1 sequence (Figure 2). For the majority of the adjacent pairs of scaffolds, the last gene of the upstream scaffold and the first gene of the downstream scaffold matched consecutive genes in the gene order of UCYN-A1 (30 cases), thereby conserving and extending the high synteny seen across the alignments. In the remaining cases, adjacent scaffold ends carried partial genes that matched different parts of the same gene in UCYN-A1 (43 partial genes in UCYN-A2 matching 21 genes in UCYN-A1).

Circular map showing all 52 scaffolds of the UCYN-A2 draft genome aligned to the UCYN-A1 chromosome. Each concentric ring represents a scaffold, with the color code representing percent nucleotide identity. The scaffolds are sorted by length, with the longest scaffold (249 164 nt) on the outermost ring, and decreasing in length towards the center ring (shortest scaffold of 675 nt). The inlet graph is a histogram of percent amino-acid identity for all 1159 ortholog genes.
Overall, the UCYN-A2 draft genome is highly similar to UCYN-A1 in gene content, synteny and basic genome features, including GC content (31%), percent of coding DNA (79.3%), codon usage (Supplementary Figure 4) and overall gene count, including two rRNA operons (Figure 2 and Table 1). There is 99% 16S rRNA gene sequence identity between both genomes. Seven RNA genes in UCYN-A2 had very similar but unannotated sequences in UCYN-A1 (91–100% nucleotide identity over 97–100% of the query sequence), and some annotated matching sequences exist in other cyanobacteria such as Calothrix sp. PCC7507 and Cyanothece sp. 8801, 8802 and 51142. These consist of one additional tRNA gene for methionine and six RNA genes annotated as noncoding RNA with unknown functions (‘other RNA genes’ in Table 1).
A total of 1159 of the 1200 UCYN-A1 proteins (Tripp et al., 2010) have closely matching sequences in UCYN-A2, that is, 96.6% of UCYN-A1’s genes are shared with UCYN-A2. For these 1159 genes, the average amino-acid sequence identity is 86.3% (range 51–100%, Figure 2). The most conserved genes (⩾95% identity) include housekeeping genes (ribosomal proteins, NADH dehydrogenase, ATP synthase), Photosystem I subunits and proteins involved in N2 fixation (nif cluster).
The previously described UCYN-A1 genome was unusual and had extensive genome reduction, lacking the genes encoding Photosystem II, RuBisCO, biosynthesis pathways for several amino acids and purines, as well as the TCA cycle and other key metabolic pathways (Zehr et al., 2008; Tripp et al., 2010). The genes missing in the UCYN-A1 genome were also absent in the UCYN-A2 draft genome. In addition to the analysis of all rejected contigs, we used TBLASTN to search the full set of unassembled sequencing reads for all 114 Cyanothece sp. 51142 genes reported missing in UCYN-A1 (Tripp et al., 2010), to test whether some of these genes might have escaped assembly. Subject reads were compared with GenBank using BLASTN against the nt database, and taxonomy was retrieved for the top 20 hits for each read. Matching reads were found for only 13 different genes out of these 114 query genes (18 total hits, incl. 5 PSII genes). Seven hits had 98–100% identity to known organisms (Synechococcus, Pelagomonas, Thalassiosira pseudonana), and four hits to an uncultured marine prokaryote. The remaining seven hits had maximal identity ranging between 79% and 89% to sequences from other organisms (Galdieria, Aureococcus, Acaryochloris, Flavobacterium, Nitrosomonas and Monosiga).
Apart from the 1159 genes shared by UCYN-A2, there are 41 UCYN-A1 genes (including 25 hypothetical proteins) that appear to be pseudogenes in UCYN-A2. These pseudogenes were either neighboring partial genes that aligned consecutively to a full open reading frame of a UCYN-A1 gene, with interrupting stop codons and/or insertions between them (a total of 21 partial genes in UCYN-A2 matching eight genes in UCYN-A1, not counting genes at scaffold ends; Table 2), or short, unannotated sequences that match only parts of UCYN-A1 genes (the remaining 33 UCYN-A1 genes). Although the evidence for pseudogenes was strong, as the UCYN-A2 sequences were from good assemblies that yielded high-coverage scaffolds, we additionally used PCR to amplify across nine random examples of these pseudogenes, confirming that the interrupting stop codons were present and were not artifacts of assembly (see Supplementary Material for details). The genome comparison revealed that such pseudogenes also exist in UCYN-A1 (Table 2).
An interesting difference between both genomes is that for all UCYN-A1 genes at least short, unannotated remnants or pseudogenes can be found in UCYN-A2, while in turn UCYN-A2 possesses 31 genes, of which 15 are hypothetical proteins, for which no traces (pseudogenes or gene remnants) were found in UCYN-A1, indicating that they have been completely lost from the genome (Table 2). The loss of these genes has in most cases resulted in further genome compaction in UCYN-A1, that is, they appear fully excised instead of being replaced by noncoding DNA (examples shown in Figure 3). The majority of these unique UCYN-A2 genes had top BLASTP similarity to genes in different Cyanothece sp. (16 genes) or in other Cyanobacteria (five genes), whereas 10 short hypothetical proteins (27–63 amino acids) had no clear phylogenetic affiliation.

Examples of missing genes in UCYN-A1, demonstrating the resulting genome compaction. A total of 31 genes was found to be unique in UCYN-A2. The alignment was done using the Artemis Comparison Tool (http://www.sanger.ac.uk/) and shows closely matching gene neighborhoods apart from the missing genes (percent nucleotide identity given for aligned genes).
In addition to interrupted genes, we note 132 genes that show differences in amino-acid length compared with orthologs in the other genome, that is, they appear truncated at either the C- or N-terminal end of the protein. For UCYN-A2, this was also confirmed for a few examples by PCR amplification (Supplementary Material). Some of these truncated genes might be pseudogenes as well. Thirteen genes in UCYN-A1 and 14 genes in UCYN-A2 had <75% of the amino acids in the comparable protein sequence in the other strain. A comparison of the ortholog pairs of UCYN-A1 and UCYN-A2 with orthologs in Cyanothece sp. 51142 showed that the truncated versions of the genes almost exclusively occur in one of the UCYN-A strains, but not in Cyanothece sp. 51142, whereas the gene length of the longer ortholog in UCYN-A1/A2 correlated well with the gene length in Cyanothece sp. 51142 (Figure 4a). Interestingly, UCYN-A1 generally possessed the shortest versions of the gene among these three genomes (Figure 4b).

(a) Comparison of amino-acid lengths of ortholog genes in UCYN-A1, UCYN-A2 and Cyanothece sp. 51142. (b) The range of percent gene length of the UCYN-A1 and UCYN-A2 orthologs compared with the Cyanothece sp. 51142 orthologs.
Overall, both genomes show extremely similar genome reduction, but there are some differences regarding which genes have become pseudogenes, and UCYN-A1 appears to have a higher level of reduction, with fully excised genes at several loci and overall greater truncation of genes. Functions affected by gene deletions or pseudogenization differ for UCYN-A1 and UCYN-A2 (Table 2), with the latter genome, for example, retaining genes involved in cell wall synthesis, vitamin import and detoxification of active oxygen species such as H2O2.
Maximum likelihood analyses confirmed that both UCYN-A strains belong to a well-supported monophyletic group of marine planktonic cyanobacteria containing Crocosphaera sp., Cyanothece sp. and other unicellular N2 fixing cyanobacteria (Sanchez-Baracaldo et al., 2014). The results of the analyses strongly support that UCYN-A2 and UCYN-A1 form a monophyletic group, that is, a sister group to Crocosphaera sp. and Cyanothece sp. (Bootstrap support 100; Figure 5). This clade of marine unicellular N2 fixers belongs to the previously described SPM group (Sanchez-Baracaldo et al., 2005) containing Synechocystis, Pleurocapsas and Microcystis (Figure 5).

Phylogeny of 57 cyanobacteria based on a concatenated alignment of 135 highly conserved protein sequences. A detailed list and description of the genes can be found in Blank and Sanchez-Baracaldo (2010). Maximum likelihood analyses were performed using RAxML 7.4.2 (Stamatakis, 2006). Bootstrap values are indicated above branches. The vertical bar marks sequences belonging to a strongly supported clade of marine unicellular N2 fixers previously described as the SPM group.
Discussion
UCYN-A is likely one of the major oceanic N2 fixers given that it has a wider geographic distribution than Trichodesmium sp., diatom symbionts or Crocosphaera sp., and can be highly abundant at certain times and places (Church et al., 2009; Moisander et al., 2010). The symbiotic relationship of UCYN-A with a eukaryotic, possibly calcifying, prymnesiophyte raises many important questions about the variability and regulation of N2 fixation in UCYN-A, the fate of the fixed nitrogen (and carbon) in the planktonic food web, the role of UCYN-A in element export to the deep ocean, and its susceptibility to ocean acidification (Thompson et al., 2012). Further, the recently recognized nifH sequence diversity in the UCYN-A clade suggests that there could be different ecotypes of UCYN-A in the ocean, which could potentially be very different in terms of genome composition and physiology (Thompson et al., 2014). The genome comparison in this study addresses this question, with the surprising discovery that both types have very similar gene content, genome reduction, but also substantially divergent DNA sequences.
UCYN-A2 has very similar gene content to UCYN-A1 and also lacks photosystem II genes, RuBisCO, TCA cycle components and other pathways. It therefore is a second, independently verified example of this kind of genome reduction in UCYN-A symbionts. Together with the highly conserved gene order, which implies gene function conservation, this suggests that UCYN-A1 and UCYN-A2 have similar functions and metabolic interactions in the symbiosis with their haptophyte hosts.
Although it can be difficult to confirm that genes are missing in unclosed genomes, we base the claim on several independent lines of evidence. (1) Many scaffolds ended with partial genes that mapped to a single UCYN-A1 gene, or ended with full genes that matched and preserved the gene order in UCYN-A1, suggesting that breaks between scaffolds were not due to missing sequence. (2) Even though there is variability in genome sequence coverage (26.7 on average, Supplementary Figure 2), it is highly unlikely that there would be no coverage at all for the long stretches of target genome needed to contain the many missing genes in UCYN-A1. (3) The rejected contigs had a GC content of 44.7% (very different from the 31% found in UCYN-A1 and UCYN-A2), sparse BLAST hits to UCYN-A1 or Cyanothece sp. (even at a very relaxed e-value threshold), and any detected hits to UCYN-A1- or Cyanothece sp.-like sequences were redundant, with genes already present in the UCYN-A2 draft genome; this ascertains that no UCYN-A2 genes were missed. (4) Searching the sequence reads by TBLASTN for all 114 Cyanothece sp. 51142 genes that appeared to be missing in UCYN-A1 returned only 13 of the query genes, of which most had highest similarity values to different organisms. (5) Recently obtained field data show peaks in nifH expression of UCYN-A2 during daytime, closely matching the temporal patterns of nifH expression determined for UCYN-A1 in the open-oligotrophic ocean around Hawaii (Church et al., 2005; Thompson et al., 2014). This may be viewed as further confirmation for the absence of oxygen-evolving PSII in UCYN-A2, given the oxygen sensitivity of the nitrogenase enzyme.
Each UCYN-A strain has only a handful of genes that are either absent or disrupted in the other genome (Table 2). The loss of genes in symbiont genomes is a gradual process, and highly reduced genomes characteristically exhibit slow gene loss in the form of erosion of individual genes or operons, rather than larger deletions via chromosomal rearrangements (Moran and Mira, 2001; Wernegreen et al., 2002; Moran, 2003). The pattern of lost, disrupted or truncated genes seen in the UCYN-A strains examined here appears consistent with such slow gene decay.
Gene inactivation and loss in symbionts mainly occurs because genes become functionally redundant and therefore non-essential, for example, due to metabolite exchange with the host. Many of the functions encoded by pseudogenes in UCYN-A1/A2 indeed appear dispensable when considered in the context of the symbiont–host relationship, such as restriction endonucleases, pyrimidine synthesis or cell motility (Table 2). However, the intact versions of those genes in the other genome, and the unique genes in UCYN-A2, raise the question whether they have been retained because their function is still important, or whether they are also non-essential/redundant but have so far escaped inactivation and elimination. Noteworthy examples are the genes involved in cell wall biogenesis and cell shape determination in UCYN-A2. The latter genes occur in rod-shaped cells and also in Cyanothece sp. 51142. These genes could indicate that UCYN-A2 has a different morphology than UCYN-A1, and could point to differences in how it is structurally associated with its host, which might also influence the fragility of the association. Interestingly, genes involved in cell wall biogenesis, which have become pseudogenes in UCYN-A1, are also among disrupted genes in the obligate cyanobacterial endosymbiont of the diatom Rhopalodia gibba (Kneip et al., 2008). Another interesting case is the UCYN-A2 peroxidase gene 2528848519. Peroxidases act in detoxifying active oxygen species such as H2O2, for example, the thioredoxin peroxidase in Synechocystis PCC6803 (68% nucleotide identity to UCYN-A2 gene) (Yamamoto et al., 1999). Active oxygen species are formed during respiration and photosynthesis, but also during many other processes (Miyake and Yokota, 2000). The presence of a peroxidase could indicate that UCYN-A2 experiences higher intracellular oxygen concentrations than UCYN-A1. UCYN-A2 would then have to respire more oxygen in order to fix N2, and in the process would generate more reactive oxygen species, thus potentially relying on this peroxidase gene.
On the basis of searches in metagenomic and metatranscriptomic data sets, the UCYN-A1 genome was initially assumed to be a global population with very similar genome sequences (⩾97% nucleotide-sequence identity, Tripp et al., 2010), analogous to the low sequence diversity seen in C. watsonii (Zehr et al., 2007; Bench et al., 2011). While the phylogenomic analysis strongly supports the two UCYN-A strains to be sister species (Figure 5), one of the striking results from our genome comparison is the relatively large range of sequence similarity seen among shared genes in UCYN-A1 and UCYN-A2 (Figure 2). The combination of this sequence divergence with the extremely high similarity in basic genome features, gene content and synteny suggests that the genome reduction occurred prior to the speciation event and genetic divergence. It is therefore likely that the common ancestor of UCYN-A1 and UCYN-A2 was already a symbiont. Vicariance might have triggered the genetic divergence in the course of speciation of the prymnesiophyte host into strains that possibly are slightly better adapted to different oceanic realms. This would have allowed the cyanobacterial genomes to accumulate gene sequence mutations after driving forces causing large genome rearrangements were no longer significant, which appears typical for symbiont genomes that have already been highly reduced (Tamas et al., 2002; Moran, 2003; Silva et al., 2003). Interestingly, genes involved in N2 fixation were among the most conserved orthologs, likely reflecting the importance of this process in maintaining the symbiosis, as it arguably represents the function most beneficial to the host and which must have been vital in the initial formation of the symbiotic relationship.
Small, conserved and highly syntenic genomes exhibiting high amino-acid divergence can also be found in the free-living heterotrophic SAR11 clade (Wilhelm et al., 2007; Grote et al., 2012). SAR 11 is an example for genome reduction due to ‘streamlining’, whereas the genome reduction seen in UCYN-A appears typical for symbiont genomes (Giovannoni et al., 2014). The amino-acid divergence between the UCYN-A strains lies within the range seen in the SAR11 Ia cluster (which have 2% 16S rRNA divergence, Grote et al., 2012). However, UCYN-A1 and UCYN-A2 have even more conserved genome content than SAR11 Ia and are considerably more conserved than members of the cyanobacterial Prochlorococcus group (Kettler et al., 2007), which appears typical for obligate intracellular organisms (Grote et al., 2012). This evolutionary pattern is unusual and suggests that the genomes of these UCYN-A strains are under strong selection, as they are highly specialized symbionts of eukaryote algae.
Although nifH sequences of UCYN-A1 and UCYN-A2 can co-occur in some samples from around the world, the question has been raised whether these two different strains could be adapted to different nutrient regimes, and could therefore have overlapping, but different distributions in the ocean (Thompson et al., 2014). However, we find no evidence in the genomes of UCYN-A1 and UCYN-A2 that would resemble genetic differentiation analogous to that in, for example, the high-light or low-light ecotypes of Prochlorococcus sp. (Moore et al., 1998; Kettler et al., 2007), or the ‘coastal’ ecotypes of Synechococcus sp. (Ahlgren and Rocap, 2006; Palenik et al., 2006). This lack of genetic differentiation, and the overall level of genome reduction, is characteristic for genomes of obligate symbionts with high dependency on their host (Moran, 2003; Hilton et al., 2013), and suggests that UCYN-A may not be directly exposed to, or affected by the external environment. Analyzing the genomes of the host algae and other UCYN-A strains will be necessary to identify genes that might represent adaptation to different environmental conditions.
While the two strains show no immediately apparent gene adaptations to cope with horizontal nutrient gradients or light quality, it is interesting that UCYN-A1 appears to be smaller than UCYN-A2 (Thompson et al., 2014), has fully excised genes compared with UCYN-A2 (Figure 3) and greater truncation of genes (Figure 4). The genomic signatures in UCYN-A point to typical genome reduction in a symbiont via genetic drift, a mechanism that is particularly enhanced under small effective population sizes (van Ham et al., 2003; Giovannoni et al., 2014). However, the further reduced genome of UCYN-A1 could also reflect an adaptation to the open ocean environment with very low levels of nutrients. Comparative genomics and ecological studies (Scanlan et al., 2009), as well as trait evolution analyses (Larsson et al., 2011), have shown a trend in genome reduction among cyanobacteria adapted to oligotrophic environments. For the host of UCYN-A, the ecological advantage of hosting a ‘diazoplast’ would come at the cost of having to sustain it with carbon energy, nutrients and a range of metabolites. Thus, it appears possible that more severe nutrient deprivation (especially for phosphorus, Scanlan et al., 2009) experienced by an open ocean ecotype of the host would also induce more extensive genome compaction (that is, streamlining) in the symbiont. Further studies are necessary to fully understand these observations.
Conclusions
The genomes of the two UCYN-A strains show considerable divergence at the amino-acid and nucleotide levels along with high conservation of genome structure, gene content and basic genome features, suggesting that they had a common symbiotic ancestor and then were separated spatially in the course of speciation. While there is some evidence for unequal distribution and possibly habitat-specific genomic streamlining in these two strains, it remains unclear whether they occupy different or overlapping niches. The genome size and the number of pseudogenes not yet fully excised from the genome of both strains might suggest that UCYN-A is still in a relatively early stage of symbiotic association with the eukaryotic host, analogous to, for example, the diazotrophic spheroid bodies found in rhopalodiacean diatoms (Kneip et al., 2008; Nakayama et al., 2014). Genome sequencing of additional UCYN-A strains and of host genomes will show whether the small differences in genetic potential reflect environmental adaptation in these organisms, and whether genetic material from UCYN-A has migrated into the host genome, as found in organelle-like stages of symbiosis (Nakayama and Ishida, 2009). The existence of different UCYN-A strains associated with different prymnesiophytes has implications for the trophic transfer and vertical export of nitrogen and carbon, and for the distribution and regulation of N2 fixation in the ocean. Further studies are needed for a better understanding of symbiotic N2 fixation and the genomic basis for UCYN-A’s role as a globally important N2 fixer.
References
Ahlgren N, Rocap G . (2006). Culture isolation and culture-independent clone libraries reveal new marine Synechococcus ecotypes with distinctive light and N physiologies. Appl Environ Microbiol. 72: 7193–7204.
Ahlgren NA, Rocap G, Chisholm SW . (2006). Measurement of prochlorococcus ecotypes using real-time polymerase chain reaction reveals different abundances of genotypes with similar light physiologies. Environ Microbiol 8: 441–454.
Bench SR, Ilikchyan IN, Tripp HJ, Zehr JP . (2011). Two strains of Crocosphaera watsonii with highly conserved genomes are distinguished by strain-specific features. Front Microbiol 2: 261.
Bergman B, Sandh G, Lin S, Larsson J, Carpenter EJ . (2013). Trichodesmium - a widespread marine cyanobacterium with unusual nitrogen fixation properties. FEMS Microbiol Rev 37: 286–302.
Blank CE, Sanchez-Baracaldo P . (2010). Timing of morphological and ecological innovations in the cyanobacteria - a key to understanding the rise in atmospheric oxygen. Geobiology 8: 1–23.
Capone DG, Zehr JP, Paerl HW, Bergman B, Carpenter EJ . (1997). Trichodesmium, a globally significant marine cyanobacterium. Science 276: 1221–1229.
Chavez FP, Pennington JT, Castro CG, Ryan JP, Michisaki RM, Schlining B et al. (2002). Biological and chemical consequences of the 1997-98 El Niño in central California waters. Progr Oceanogr 54: 205–232.
Church MJ, Mahaffey C, Letelier RM, Lukas R, Zehr JP, Karl DM . (2009). Physical forcing of nitrogen fixation and diazotroph community structure in the North Pacific subtropical gyre. Global Biogeochem Cycles 23: GB2020.
Church MJ, Short CM, Jenkins BD, Karl DM, Zehr JP . (2005). Temporal patterns of nitrogenase gene (nifH) expression in the oligotrophic North Pacific Ocean. Appl Environ Microbiol 71: 5362–5370.
Diez B, Bergman B, Pedros-Alio C, Anto M, Snoeijs P . (2012). High cyanobacterial nifH gene diversity in Arctic seawater and sea ice brine. Environ Microbiol Rep 4: 360–366.
Dugdale RC, Menzel DW, Ryther JH . (1961). Nitrogen fixation in the Sargasso Sea. Deep-Sea Research 7: 297–300.
Foster RA, Kuypers MMM, Vagner T, Paerl RW, Musat N, Zehr JP . (2011). Nitrogen fixation and transfer in open ocean diatom-cyanobacterial symbioses. ISME J 5: 1484–1493.
Foster RA, Zehr JP . (2006). Characterization of diatom-cyanobacteria symbioses on the basis of nifH, hetR and 16S rRNA sequences. Environ Microbiol 8: 1913–1925.
Giovannoni SJ, Thrash JC, Temperton B . (2014). Implications of streamlining theory for microbial ecology. ISME J 8: 553–565 doi:10.1038/ismej.2014.60.
Glibert PM, Bronk DA . (1994). Release of dissolved organic nitrogen by marine diazotrophic cyanobacteria Trichodesmium spp. Appl Environ Microbiol 11: 3996–4000.
Grote J, Thrash C, Huggett MJ, Landry ZC, Carini P, Giovannoni SJ . (2012). Streamlining and core genome conservation among highly divergent members of the SAR11 clade. mBio 3: e00252–12.
Hilton JA, Foster RA, Tripp JH, Carter BJ, Zehr JP, Villareal TA . (2013). Genomic deletions disrupt nitrogen metabolism pathways of a cyanobacterial diatom symbiont. Nat Commun 4: 1767.
Janson S, Wouters J, Bergman B, Carpenter EJ . (1999). Host specificity in the Richelia-diatom symbiosis revealed by hetR gene sequence analysis. Environ Microbiol 1: 431–438.
Johnson ZI, Zinser ER, Coe A, McNulty NP, Woodward EMS, Chisholm SW . (2006). Niche partitioning among Prochlorococcus ecotypes along ocean-scale environmental gradients. Science 311: 1737–1740.
Karl D, Letelier R, Tupas L, Dore J, Christian J, Hebel D . (1997). The role of nitrogen fixation in biogeochemical cycling in the subtropical North Pacific Ocean. Nature 388: 533–538.
Kettler GC, Martiny AC, Huang K, Zucker J, Coleman ML, Rodrigue S et al. (2007). Patterns and implications of gene gain and loss in the evolution of Prochlorococcus. PLoS Genet 3: e231.
Kneip C, Vobeta C, Lockhart P, Maier U . (2008). The cyanobacterial endosymbiont of the unicellular algae Rhopalodia gibba shows reductive genome evolution. BMC. Evol Biol 8: 30.
Langlois RJ, Hummer D, LaRoche J . (2008). Abundances and distributions of the dominant nifH phylotypes in the Northern Atlantic Ocean. Appl Environ Microbiol 74: 1922–1931.
Larsson J, Nylander J, Bergman B . (2011). Genome fluctuations in cyanobacteria reflect evolutionary, developmental and adaptive traits. BMC. Evol Biol 11: 187.
Miyake C, Yokota A . (2000). Determination of the rate of photoreduction of O2 in the water-water cycle in watermelon leaves and enhancement of the rate by limitation of photosynthesis. Plant Cell Physiol 41: 335–343.
Moisander PH, Beinart RA, Hewson I, White AE, Johnson KS, Carlson CA et al. (2010). Unicellular cyanobacterial distributions broaden the oceanic N2 fixation domain. Science 327: 1512–1514.
Moore LR, Rocap G, Chisholm SW . (1998). Physiology and molecular phylogeny of coexisting Prochlorococcus ecotypes. Nature 393: 464–467.
Moran NA . (2003). Tracing the evolution of gene loss in obligate bacterial symbionts. Curr Opin Microbiol 6: 512–518.
Moran NA, Mira A . (2001). The process of genome shrinkage in the obligate symbiont Buchnera aphidicola. Genome Biol 2: RESEARCH0054.
Mulholland MR . (2007). The fate of nitrogen fixed by diazotrophs in the ocean. Biogeosciences 4: 37–51.
Mulholland MR, Bernhardt PW, Blanco-Garcia JL, Mannino A, Hyde K, Mondragon E et al. (2012). Rates of dinitrogen fixation and the abundance of diazotrophs in North American coastal waters between Cape Hatteras and Georges Bank. Limnol Oceanogr 57: 1067–1083.
Nakayama T, Ishida K-i . (2009). Another acquisition of a primary photosynthetic organelle is underway in Paulinella chromatophora. Curr Biol 19: R284–R285.
Nakayama T, Kamikawa R, Tanifuji G, Kashiyama Y, Ohkouchi N, Archibald JM et al. (2014). Complete genome of a nonphotosynthetic cyanobacterium in a diatom reveals recent adaptations to an intracellular lifestyle. Proc Natl Acad Sci USA 111: 11407–11412.
Needoba JA, Foster RA, Sakamoto C, Zehr JP, Johnson KS . (2007). Nitrogen fixation by unicellular diazotrophic cyanobacteria in the temperate oligotrophic North Pacific Ocean. Limnol Oceanogr 52: 1317–1327.
Palenik B, Ren QH, Dupont CL, Myers GS, Heidelberg JF, Badger JH et al. (2006). Genome sequence of Synechococcus CC9311: Insights into adaptation to a coastal environment. ProcNatlAcad Sci USA 103: 13555–13559.
Partensky F, Blanchot J, Vaulot D . (1999). Differential distribution and ecology of Prochlorococcus and Synechococcus in oceanic water: a review. Bull Inst Oceanogr Special 19: 457–475.
Rees AP, Gilbert JA, Kelly-Gerreyn BA . (2009). Nitrogen fixation in the western English Channel (NE Atlantic Ocean). Mar Ecol Prog Ser 374: 7–12.
Ruby JG, Bellare P, DeRisi JL . (2013). PRICE: software for the targeted assembly of components of (meta) genomic sequence data. G3: (Bethesda) 3: 865–880.
Sanchez-Baracaldo P, Ridgwell A, Raven JA . (2014). A neoproterozoic transition in the marine nitrogen cycle. Curr Biol 24: 652–657.
Sanchez-Baracaldo P, Hayes PK, Blank CE . (2005). Morphological and habitat evolution in the cyanobacteria using a compartmentalization approach. Geobiology 3: 145–165.
Scanlan DJ . (2003). Physiological diversity and niche adaptation in marine Synechococcus. In Advances in Microbial Physiology Vol. 47. Academic Press Ltd: London, pp 1–64.
Scanlan DJ, Ostrowski M, Mazard S, Dufresne A, Garczarek L, Hess WR et al. (2009). Ecological genomics of marine picocyanobacteria. Microbiol Mol Biol Rev. 73: 249–299.
Scanlan DJ, West NJ . (2002). Molecular ecology of the marine cyanobacterial genera Prochlorococcus and Synechococcus. FEMS Microbiol Ecol 40: 1–12.
Scharek R, Tupas L, Karl DM . (1999). Diatom fluxes to the deep sea in the oligotrophic North Pacific gyre at Station ALOHA. Mar Ecol Prog Ser 182: 55–67.
Short SM, Zehr JP . (2005). Quantitative analysis of nifH genes and transcripts from aquatic environments. In: Jared RL, (eds) Methods Enzymology Vol. 397. Academic Press, pp 380–394.
Silva FJ, Latorre A, Moya A . (2003). Why are the genomes of endosymbiotic bacteria so stable? Trends genet 19: 176–180.
Sohm JA, Webb EA, Capone DG . (2011). Emerging patterns of marine nitrogen fixation. Nat Rev Microbiol 9: 499–508.
Stal LJ . (2009). Is the distribution of nitrogen-fixing cyanobacteria in the oceans related to temperature? Environ Microbiol 11: 1632–1645.
Stamatakis A . (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690.
Tamas I, Klasson L, Canback B, Naslund AK, Eriksson AS, Wernegreen JJ et al. (2002). 50 million years of genomic stasis in endosymbiotic bacteria. Science 296: 2376–2379.
Thompson AW, Carter BJ, Turk-Kubo KA, Malfatti F, Azam F, Zehr JP . (2014). Genetic diversity of the unicellular nitrogen-fixing cyanobacteria UCYN-A and its prymnesiophyte host. Environ Microbiol doi:10.1111/1462-2920.12490.
Thompson AW, Foster RA, Krupke A, Carter BJ, Musat N, Vaulot D . (2012). Unicellular cyanobacterium symbiotic with a single-celled eukaryotic alga. Science 337: 1546–1550.
Tripp HJ, Bench SR, Turk KA, Foster RA, Desany BA, Niazi F et al. (2010). Metabolic streamlining in an open ocean nitrogen-fixing cyanobacterium. Nature 464: 90–94.
van Ham RC, Kamerbeek J, Palacios C, Rausell C, Abascal F, Bastolla U et al. (2003). Reductive genome evolution in Buchnera aphidicola. Proc Natl Acad Sci USA 100: 581–586.
Villareal TA . (1992). Marine nitrogen-fixing diatom - cyanobacteria symbioses. In: Carpenter EJ, Capone DG, Rueter JG, (ed) Marine Pelagic Cyanobacteria: Trichodesmium and Other Diazotrophs. Kluwer Academic Publishers: The Netherlands, pp 163–175.
Voss M, Bange HW, Dippner JW, Middelburg JJ, Montoya JP, Ward B . (2013). The marine nitrogen cycle: recent discoveries, uncertainties and the potential relevance of climate change. Philos TransR Soc B 368: 20130121–20130121.
Wernegreen JJ, Lazarus AB, Degnan PH . (2002). Small genome of Candidatus Blochmannia, the bacterial endosymbiont of Camponotus, implies irreversible specialization to an intracellular lifestyle. Microbiology 148: 2551–2556.
Wilhelm LJ, Tripp HJ, Givan SA, Smith DP, Giovannoni SJ . (2007). Natural variation in SAR11 marine bacterioplankton genomes inferred from metagenomic data. Biol Direct 2: 27.
Yamamoto H, Miyake C, Dietz K-J, Tomizawa K-I, Murata N, Yokota A . (1999). Thioredoxin peroxidase in the Cyanobacterium Synechocystis sp. PCC 6803. FEBS Lett 447: 269–273.
Zehr JP, Bench SR, Carter BJ, Hewson I, Niazi F, Shi T et al. (2008). Globally distributed uncultivated oceanic N2-fixing cyanobacteria lack oxygenic photosystem II. Science 322: 1110–1112.
Zehr JP, Bench SR, Mondragon EA, McCarren J, DeLong EF . (2007). Low genomic diversity in tropical oceanic N2-fixing cyanobacteria. Proc Natl Acad Sci USA 104: 17807–17812.
Zehr JP, Kudela RM . (2011). Nitrogen cycle of the open ocean: from genes to ecosystems. Annu Rev Mar Sci 3: 197–225.
Zehr JP, Mellon MT, Zani S . (1998). New nitrogen fixing microorganisms detected in oligotrophic oceans by the amplification of nitrogenase (nifH) genes. Appl Environ Microbiol 64: 3444–3450.
Zehr JP, Paerl HW . (2008). Molecular ecological aspects of nitrogen fixation in the marine environment. In: Kirchman DL, (ed) Microbial Ecology of the Oceans. Wiley-Liss, Inc.: Durham, NC, pp 481–525.
Zehr JP, Waterbury JB, Turner PJ, Montoya JP, Omoregie E, Steward GF et al. (2001). Unicellular cyanobacteria fix N2 in the subtropical North Pacific Ocean. Nature 412: 635–638.
Acknowledgements
We thank F Malfatti and F Azam at SIO for assistance with sampling and for providing lab facilities, Kendra Turk-Kubo for carrying out PCR reactions to confirm pseudogene sequences, S Biller at MIT for assistance with sample handling, S Bench for bioinformatics assistance, and A Thompson, J Tripp and J Hilton for valuable discussions. Comments from three anonymous reviewers greatly improved the paper. This work was supported by a Gordon and Betty Moore Foundation Marine Investigator Award (JZ), the MEGAMER facility (supported by GBMF) and the Center for Microbial Oceanography: Research and Education (NSF grant 0424599).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Bombar, D., Heller, P., Sanchez-Baracaldo, P. et al. Comparative genomics reveals surprising divergence of two closely related strains of uncultivated UCYN-A cyanobacteria. ISME J 8, 2530–2542 (2014). https://doi.org/10.1038/ismej.2014.167
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2014.167
This article is cited by
-
Symbiotic UCYN-A strains co-occurred with El Niño, relaxed upwelling, and varied eukaryotes over 10 years off Southern California
ISME Communications (2023)
-
Distribution and survival strategies of endemic and cosmopolitan diazotrophs in the Arctic Ocean
The ISME Journal (2023)
-
Unusual marine cyanobacteria/haptophyte symbiosis relies on N2 fixation even in N-rich environments
The ISME Journal (2020)
-
Evolutionary dynamics of the chromatophore genome in three photosynthetic Paulinella species
Scientific Reports (2019)
-
Small eukaryotic phytoplankton communities in tropical waters off Brazil are dominated by symbioses between Haptophyta and nitrogen-fixing cyanobacteria
The ISME Journal (2018)
