
Abstract
Symbioses are increasingly seen as dynamic ecosystems with multiple associates and varying fidelity. Symbiont specificity remains elusive in one of the most ecologically successful and economically damaging eukaryotic symbioses: the ambrosia symbiosis of wood-boring beetles and fungi. We used multiplexed pyrosequencing of amplified internal transcribed spacer II (ITS2) ribosomal DNA (rDNA) libraries to document the communities of fungal associates and symbionts inside the mycangia (fungus transfer organ) of three ambrosia beetle species, Xyleborus affinis, Xyleborus ferrugineus and Xylosandrus crassiusculus. We processed 93 beetle samples from 5 locations across Florida, including reference communities. Fungal communities within mycangia included 14–20 fungus species, many more than reported by culture-based studies. We recovered previously known nutritional symbionts as members of the core community. We also detected several other fungal taxa that are equally frequent but whose function is unknown and many other transient species. The composition of fungal assemblages was significantly correlated with beetle species but not with locality. The type of mycangium appears to determine specificity: two Xyleborus with mandibular mycangia had multiple dominant associates with even abundances; Xylosandrus crassiusculus (mesonotal mycangium) communities were dominated by a single symbiont, Ambrosiella sp. Beetle mycangia also carried many fungi from the environment, including plant pathogens and endophytes. The ITS2 marker proved useful for ecological analyses, but the taxonomic resolution was limited to fungal genus or family, particularly in Ophiostomatales, which are under-represented in our amplicons as well as in public databases. This initial analysis of three beetle species suggests that each clade of ambrosia beetles and each mycangium type may support a functionally and taxonomically distinct symbiosis.
Similar content being viewed by others


Introduction
Some symbioses conform to the traditional one-on-one mode of symbiotic relationships, but it is increasingly understood that many symbioses are more open and consist of multiple associates with varying degree of fidelity (Caldera et al., 2009). This is particularly true for symbioses between animals and microbes (Gilbert et al., 2012). The improved understanding is partly a result of the availability of high-throughput, culture-independent techniques of community surveys and partly a result of maturation of the field of symbiology. At the same time, it is becoming clear that not all microbes discoverable within animals and plants by modern techniques are symbionts. Contemporary understanding of symbioses emphasizes the shared evolutionary and ecological dynamics of communities where cooperation among members prevails over antagonism (Douglas, 2010).
One of the most common symbioses in any forest ecosystem occurs between wood-boring insects and fungi. The two partners have complementary qualities: the insects fly and can mechanically colonize wood, while their fungal partners concentrate nutrients from the plant tissue and deliver them to the insect in the form of specialized mycelium. Wood is one of the world’s largest stores of biomass, making this symbiosis one of the most ecologically successful and economically important. The most advanced stage of this relationship is the farming of ambrosia fungi by ambrosia beetles. This obligate mutualism evolved multiple times between wood-boring beetles (Curculionidae, Scolytinae and Platypodinae) and several groups of fungi, particularly Ophiostomatales, Microascales, Hypocreales and Saccharomycotina (Beaver, 1989; Farrell et al., 2001; Massoumi Alamouti et al., 2009).
The scolytine beetle–fungus symbiosis is receiving an increasing amount of research attention because of the massive impact, both ecological and economic, that these organisms can cause. For example, the largest outbreak of forest pests in recorded history is currently occurring in the northern and western regions of North America, caused by several Dendroctonus bark beetles closely associated with symbiotic fungi. Also, new introductions of ambrosia beetles and fungi into non-native regions are increasingly common; invasive ambrosia insect–fungus couples are eradicating naive, native tree species in many places around the world (Hulcr and Dunn, 2011).
Recent research on the ambrosia symbiosis illuminated a relationship that is more species-rich, more ecologically dynamic and more evolutionarily promiscuous than any other insect–fungus symbiosis. The obligate ambrosia symbiosis is now known to have evolved at least eleven times in insects and seven times in fungi (Van der Walt, 1972; Hulcr et al., 2007; Massoumi Alamouti et al., 2009; Kasson et al., 2013) and involved origins of many different fungus transmission organs (mycangia). Still, one of the essential characteristics of the symbiosis—specificity—remains controversial. It has been traditionally assumed that ambrosia beetles are mostly associated with a single dominant fungus (Hubbard, 1896, 1897; Doane and Gilliland, 1929; Leach et al., 1940; Francke-Grosmann, 1956; Batra, 1963), but an increasing number of studies suggests that some beetle species may have more than one associate (Batra, 1966; Norris, 1979; Kühnholz, 2004; Gebhardt et al., 2004; Harrington et al., 2010; Carrillo et al., 2014). Recent experiments using cultured ambrosia fungi suggested that sympatric communities of ambrosia beetles may actually share a large pool of fungi and that horizontal symbiont exchange may be common (Carrillo et al., 2014).
The reasons for the unsettled understanding of beetle-fungus specificity have involved a range of technological obstacles. Aside from the minute size and cryptic nature of both the beetles and the fungi, the main obstacle for culture-independent approaches has been that the most commonly used marker for fungal community surveys—the internal transcribed spacer (ITS) rDNA region—is difficult to use in many ambrosia fungus groups. This region is extremely GC-rich and difficult to amplify and sequence in many Ophiostomatales (Fraedrich et al., 2008). In other groups, such as Ambrosiella (Microascales), ITS sequences are severely under-represented or incorrectly identified in public sequence databases, or ITS rDNA is highly conserved and uninformative in species identification (e.g., Fusarium (Hypocreales) (O’Donnell, 2000)). Another obstacle, important for both culturing and culture-independent approaches, has been the spatial distribution of fungal associates in and on the beetle body. The beetles live in a very fungus-rich environment, but the coevolved symbionts occur almost exclusively within the beetle mycangia and in parts of the galleries made by the beetles in wood and for only a limited part of the beetle life cycle (Kajimura and Hijii, 1992). Thus the traditional method of analyzing fungal communities from entire macerated beetle bodies has introduced a large number of non-symbionts into the literature and a large uncertainty regarding the true associates.
In this work, we have carried out the first high-throughput survey of communities of fungi associated with mycangia of three sympatric ambrosia beetles. Beetle species were chosen to enable tests of several hypotheses. We included two clades with different modes of fungus transportation (different mycangia), and species that commonly occupy the same dead trees throughout the southeastern United States of America. To allow for strong statistical inference, the sampling included sufficient numbers of individuals and those were analyzed with a high-throughput sequence-based community survey method.
Specifically, we tested whether ambrosia beetle mycangia contain one, or a very few, faithful fungal symbionts or whether they are more permissive. This test also implicitly addressed the question whether a culture-independent approach would corroborate the taxonomic identities of ambrosia fungi previously inferred from culture-based studies.
We also examined new questions that have not been addressed in previous publications. For example, the total diversity of the mycangial communities has not yet been quantified and may have been underestimated by culturing methods. Quantitative community data also allowed us to test fundamental symbiology questions, such as whether the mycangial communities fit the pattern of high-fidelity exclusive symbioses, or the pattern of loose assemblages of species with varying degrees of association. Finally, we were able to test whether fungi are specific to geographic localities or whether they maintain specificity to their beetle species across localities.
To answer those questions, we provided the first fine-scale and high-throughput data on fungal communities in ambrosia beetle mycangia. We comprehensively characterized communities of fungal associates on multiple levels: within beetle species with sufficient local sampling, within species across a larger region and also between three species.
Materials and methods
Beetle species and locality selection
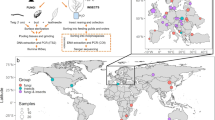
We sampled three ambrosia beetle species commonly found across the southeastern United States: Xylosandrus crassiusculus, Xyleborus affinis, and Xyleborus ferrugineus. This selection has multiple advantages for this and future work. For example, these species represent two types of mycangia (Figure 1). The mandibular mycangium (X. affinis and X. ferrugineus) has openings in the oral cavity and is the ancestral and most widely distributed mycangium type in the beetle tribe Xyleborini (Hulcr and Cognato, 2010; Cognato et al., 2011). The mesonotal mycangium (X. crassiusculus) is located between the pronotum and the elytral basis; it is larger and more recently evolved (Hulcr and Cognato, 2010; Cognato et al., 2011). Another advantage of this selection of taxa is that all three species are nearly globally distributed, spread partly through human trade (Wood and Bright, 1992). X affinis and X. ferrugineus are native in warm and humid regions throughout the Americas, but both have now colonized the tropics and subtropics of other continents. X. crassiusculus is native to eastern Asia and is aggressively colonizing the rest of the world, frequently becoming a dominant ambrosia beetle in new regions. This history of range expansion makes these species ideal candidates for future analyses of symbiont fidelity during biological invasions.

(a) Sampling location in Florida, US. Pie size corresponds to the relative number of beetle specimens collected from each location; color sections indicate ratios of three beetle species. (b) Sampling locations on the beetles: Digital cross-sections of Xyleborus ferrugineus and Xylosandrus crassiusculus showing the two different types of mycangia, mandibular and mesonotal, respectively.
All three species were sampled from various habitats across Florida (Figure 1, Supplementary Table S1). Effort was made to collect all three species from most of the locations, which enabled us to compare the effect of locality as well as beetle species on the fungal community. Altogether, 93 samples were processed, including 35 X. crassiusculus from 4 locations, 38 X. affinis from 2 locations and 20 X. ferrugineus from 3 locations. To test for contamination, PCR biases and sequencing biases, several control samples were included: a negative control (a blank sample passed through all steps of library preparation from DNA extraction to sequencing) and several reference communities of pure culture fungal strains previously isolated from the same beetle species from the same locations (Supplementary Table S1).
Beetles were collected during their dispersal flight, which is when the mycangium contains the largest amount of symbiont inoculum (Kajimura and Hijii, 1992). Beetles were collected using plastic 2-liter bottles with a cut-out window, filled with approximately 50 ml of ethanol. Ethanol served as both the bait and the preservation medium. Traps were checked daily, and all trapped beetles were stored in 95% ethanol and at −80 °C.
DNA extraction
Before the extraction of fungi from mycangia, beetles were surface-washed by 15-s vortex in sterile phosphate buffer saline with 0.1% Tween, followed by 5 s vortex in 40% ethanol. After drying, beetles were either stored in 95% ethanol at −80 °C or immediately processed for mycangial extraction. The contents of the mesonotal mycangia of X. crassiusculus were extracted from beetles stretched with 0.15-mm minuten pins on sterilized paraffin under a dissecting microscope. The fungal mass was removed with a pin and immediately placed in extraction buffer. In X. affinis and X. ferrugineus, the content of the mandibular mycangia cannot be accessed directly; instead the frontal part of the head that contains the paired mycangium was cut off, avoiding the esophagus, and crushed. Fungal DNA was extracted with the UltraClean Microbial DNA (MoBio, Carlsbad, CA, USA) isolation kit according to manufacturer’s instructions. DNA concentrations were assessed using the PicoGreen fluorescent dye in the Quant-iT kit (Life Technologies, Carlsbad, CA, USA) on a Realplex cycler (Eppendorf, Hauppauge, NY, USA) and adjusted to the concentration of 10 ng μl−1. The presence of amplifiable DNA in all the samples was confirmed by a test amplification.
Marker selection
The marker used for the fungal community survey was the ITS2 of the nuclear rDNA, on average 180 bp long. The ITS2 marker provides several benefits: (1) The ITS1-5.8S-ITS2 part of the nuclear ribosomal DNA (ITS) has been proposed as the universal ‘barcode’ for identification of fungi (Schoch et al., 2012); (2) As the fungal ‘barcode’, ITS as a whole and ITS2 in particular is the best represented fungal genomic element in public databases (Nilsson et al., 2009); (3) The length of ITS2 is less variable than the whole ITS region. This is important, as length variation can bias community pyrosequencing toward shorter amplicons (Ihrmark et al., 2012); (4) Using just the ITS2 region instead of the entire ITS region eliminates the formation of chimeric amplicons of heterospecific ITS1 and ITS2 regions, induced by the annealing of the highly conservative 5.8S regions from different species (Quince et al., 2009; Bellemain et al., 2010). To further increase taxonomic coverage of the primers, a recently developed degenerate forward primer gITS7 was used (5′-GTGARTCATCGARTCTTTG-3′) (Ihrmark et al., 2012) in combination with a universal ITS4 reverse primer (5′-TCCTCCGCTTATTGATATGC-3′) (White et al., 1990).
Using ITS as a marker for an ambrosia community survey also has its problems. It is known that several traditionally used primers for the ITS region in fungal community surveys (that is, ITS1f and ITS4; Jumpponen and Jones, 2009; Amend et al., 2010; Arfi et al., 2012) amplify poorly or not at all in many Ophiostomatales, including several clades of ambrosia fungi (Fraedrich et al., 2008). The amplification failure is likely due to Ophiostomatales-specific formation of secondary structures in the GC-rich region at the conservative end of the 18S nrDNA where ITS1f and other 5′ primers anneal (White et al., 1990; Gardes and Bruns, 1993). Fortunately, during our trials with a high-performance polymerase (Q5 High-Fidelity DNA polymerase, New England Biolabs, Ipswich, MA, USA), the amplification of ITS2 was reliable (see below). With the two above-mentioned primers, we have successfully amplified ITS2 from DNA extracts from pure cultures that included representatives of three clades of common ambrosia fungi, such as several Raffaelea spp. (Ophiostomatales).
PCR amplification and library preparation
The marker was amplified in all samples following a nested-PCR approach. First, the total mycangial DNA was subjected to amplification with the template-specific primers gITS7 and ITS4 for 25 cycles. The number of cycles was lower than usual to prevent depletion of some of the forward primers in the degenerate mixture. All samples were amplified in triplicate to decrease stochastic variability between reactions (Schmidt et al., 2013). Pooled PCR reactions were cleaned by UltraClean PCR clean-up kit (MoBio). Second, a 1-μl aliquot of this amplicon was subjected to a 10-cycle amplification with fusion primers. The forward fusion primer gITS7 consisted of the template primer, the pyrosequencing adapter A and a 10-bp sample-specific barcode. Barcodes were designed, and fusion primers were tested using EDITTAG (Faircloth and Glenn, 2012). The ‘edit distance’ metric was conservatively set to five to minimize sample assignment errors due to sequencing, replication and oligonucleotide synthesis errors. An edit distance of 5 means that at least 5 insertions, substitutions or deletions would have to occur in one tag to convert it into another tag and therefore confuse demultiplexing. For barcode sequences of all samples, see Supplementary Table S1. The reverse ITS4 fusion primer consisted of the template primer and the pyrosequencing adapter B.
This nested PCR process reduces amplification bias caused by applying long fusion primers directly to the template DNA and results in the recovery of higher taxon diversity (Berry et al., 2011). Amplification was conducted on Mastercycler Nexus (Eppendorf) in 25-μl reactions with proofreading Q5 High-Fidelity polymerase (New England Biolabs) at 0.02 U μl−1 in Q5 reaction buffer, 2 mM MgCl2, 200 μM dNTPs, 500 nM gITS7 primer, 300 nM ITS4 primer and 10 ng of template. Cycling conditions were as follows: initial denaturation at 95 °C for 4 min followed by 25 cycles of (50 s at 94 °C, 50 s at 56 °C, 45 s at 72 °C) and a final elongation step for 10 min at 72 °C.
The second-step PCR products were individually cleaned up (UltraClean PCR clean-up kit, MoBio), their concentrations measured using the PicoGreen-based Quant-iT kit (Life Technologies), and pooled in equimolar proportions to create an amplicon library at a final concentration of 10 ng μl−1. The pooled amplicon library was cleaned up again using UltraClean PCR clean-up kit (MoBio). The library was sequenced on two 1/8th regions of a sequencing plate on GS-FLX Titanium (454 Life Sciences, Branford, CT, USA) at the Interdisciplinary Center for Biotechnology Research at the University of Florida. We expected to obtain an average sequencing depth of 2000 reads per sample provided that standard yield per region is 100 000 reads. Raw demultiplexed sequencing data with sample annotations are available at the Short Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/) under the study accession number SRX348622.
Quality control and reference communities
To confirm that our PCR-based results do not suffer from major biases and that they reflect the fungal communities within the beetles, we included three quality-control measures.
First, we incorporated two reference communities of known composition into the sequencing library. One was composed of several representatives of genus Raffaelea and the second one was composed of representatives of clades previously reported as ambrosia fungi, including Ophiostomatales, Microascales and a basidiomycetous yeast.
Second, to assure that major culturable symbionts with known PCR biases are not missed (for example, Raffaelea, Fraedrich et al., 2008), the sequencing output was compared with a reference culture data set. Relative abundances of dominant fungal taxa were estimated by dilution-to-extinction plating of mycangia on Potato Dextrose Agar media from mycangia of the same beetle species (Supplementary Table S6).
Third, we exhaustively reviewed primary literature and extracted records of fungal associates isolated from X. affinis, X. ferrugineus and X. crassiusculus (Table 1). Due to taxonomic uncertainties in older literature, we only surveyed reports published after the first reclassification of ambrosia fungi by Batra (1967).
Data processing
Sequencing data were processed using QIIME 1.7.0 (Caporaso et al., 2010). Reads <150 bp, >600 bp, with >6 ambiguous bases and with homopolymers >10 bp, were discarded. The minimal quality score was set to 25, and no mismatches in the primer sequence were allowed. Data were denoised according to Reeder and Knight (2010). Chimeric sequences were removed using a combination of reference-based and de novo chimera detection with USEARCH 6.1 (Edgar et al., 2011); only reads detected as chimeric with both approaches were discarded. For the subsequent analyses, the ITS2 region was extracted from each sequence using Fungal ITS extractor (Nilsson et al., 2010), as the conserved flanking regions are known to distort similarity searches, taxonomic assignments and clustering results (Bruns and Shefferson, 2004). The resulting ITS2 reads were clustered to operational taxonomic units (OTUs) by UCLUST (Edgar, 2010), with 97% similarity threshold to reference sequences. As the reference database, we used the UNITE+INSDC data sets, accessed 26 August 2013 (Abarenkov et al., 2010). The reference sequences represented species hypotheses (Koljalg et al., 2013) defined by dynamic clustering with a floating level of similarity recommended by specialists on particular lineages of fungi. Reads that failed to hit an existing reference sequence were clustered de novo. Singletons (clusters containing only one read) were discarded as they are indistinguishable from sequencing errors (Kunin et al., 2010). For each OTU, the most abundant read was designated as the representative sequence. OTUs were identified taxonomically using RDP classifier (minimum confidence set to 0.8) (Wang et al., 2007) against the UNITE+INSDC data set.
Statistical analysis
Statistical analysis was conducted in QIIME 1.7.0 (Caporaso et al., 2010) unless noted otherwise. For most analyses, samples were rarefied to the number of reads in the least numerous sample, which had 670 reads. The completeness of fungal taxon sampling was estimated by rarefaction curves.
Alpha diversity of fungal communities in each of the three beetle species was estimated by species accumulation curves, the Chao1 estimator of total diversity, the Dominance index (1−Simpson index) and the Shannon index. To quantify differences in fungal communities between samples (beta diversity), a matrix of pairwise Bray–Curtis dissimilarities was computed. The non-phylogenetic Bray–Curtis metric was used, as the ITS2 region is unalignable between different fungal genera. Sample similarities were visualized using Principal Coordinate Analysis. The effect of beetle species, location and their interaction on the fungal composition of samples was tested using two complementary nonparametric approaches: analysis of similarities (ANOSIM; Clarke, 1993) and permutational multivariate analysis of variance (PERMANOVA; Anderson, 2001). Statistical significance was determined with 999 permutations. The effect of locality and the interaction between locality and beetle species was tested using the two largest samples (Colt Creek and Lake Wales) owing to unbalanced species representation in other locations.
To test if any particular OTU was significantly associated with any particular beetle species, ANOVA and the G-test of independence were used. The G-test is used for presence/absence data, whereas ANOVA is used for relative abundance data. Both tests employed 100 de novo rarefied OTU tables so the resulting P-values are the averages of P-values from single tests. The significance level for ANOVA was corrected for multiple comparisons by the false discovery rate correction (Benjamini and Hochberg, 1995).
Identification of core associates
The fungal community of each beetle species was partitioned into core OTUs (persistent, abundant, non-randomly associated and likely biologically relevant taxa) and occasional or rare OTUs (opportunist, contaminants or other non-symbionts). Completely sampled biological communities typically follow log-normal species abundance distribution (SAD) (Magurran and Henderson, 2003). Culture-independent high-throughput data sets, however, are frequently negatively skewed, which is thought to be caused by low-frequency extraneous species (Unterseher et al., 2011). We identified core fungal taxa within each beetle species based on discontinuity in a log-normal SAD using persistence-abundance plots. First, OTUs were plotted on a persistence-abundance plot (Magurran and Henderson, 2003) so that the number of samples in which each taxon was observed was plotted against the maximum abundance of each taxon in any one sample (Unterseher et al., 2011). The plots provide means for visual detection of a discontinuity; OTUs are iteratively separated in two and separately tested for fit with the log-normal SAD (core species, right) and log-series SAD (rare or transient species, left).
We used both incidence (presence/absence) and abundance of reads. PCR-based community surveys are sometimes summarized using only incidence data to circumvent biases in DNA extraction and PCR (Bellemain et al., 2010). On the other hand, incidence-only data may overemphasize environmental contaminants that are frequent but have minor abundance and have no role in the symbiosis. Also, we used amplification of short fragments and degenerate primers with large taxonomic coverage, both of which were shown to improve reconstruction of true community structure (Ihrmark et al., 2012).
Results and discussion
Data
In total, 197 672 reads were generated. After quality filtering, denoising and chimera removal, 148 923 reads were retained for further analysis. After demultiplexing (assignment of reads to beetle samples), the average read count per sample was 1551. The sequencing depth was relatively even across samples: the poorest sample contained 670 reads and the richest sample contained 4000 reads. The 97% sequence similarity cutoff produced 402 clusters representing OTUs with at least two reads. This sequencing depth was sufficient for recovering most of the diversity present in each beetle species given the similarity threshold, as indicated by the flattening of species accumulation curves (Supplementary Figure S1).
Symbiont community diversity
Our results show an order of magnitude greater diversity of fungal species occurring in beetle mycangia than previously reported from culturing studies (Table 1). The estimated total numbers of associates within each beetle species were similar: the Chao1 estimate of total diversity ranged from 14.71 to 20.06 (Table 2). These estimates were likely approaching the true diversity, as our sampling was near exhaustive (Supplementary Figure S1). The estimates may have been slightly underestimated as singleton reads were removed during quality control, but singletons are probably not relevant in the symbiosis.
Even though ambrosia beetle genera have similar overall diversity of associated fungi, it appears that each genus, characterized by a unique type of mycangium, hosts communities of different taxonomic composition and different diversity structure (Figure 2, Table 2, Supplementary Table S2, Supplementary Figure S1). The two representatives of the beetle genus Xyleborus with mandibular mycangia (X. affinis and X. ferrugineus) had fungal communities similar in their diversity and evenness (community dominance index=0.19 and 0.23, respectively; Figure 2, Table 2). On the other hand, fungal communities of Xylosandrus crassiusculus were heavily dominated (Figure 2, dominance index=0.49) by a single species, Ambrosiella sp., and also contained more incidental transient species.

Stack plot showing species richness and evenness of mycangial fungal communities of the three beetle species. Upper stacks report averages within each species, lower stacks report communities in each individual. Each color band represents different fungal OTU; the band width corresponds to the relative abundance of reads of the OTU in the data set. Arrows point out the two different types of mycangia.
This study analyzed only a single representative of the mesonotal mycangium; however, the differences were significant and also reflect our reference cultures and data in the literature (Table 1). Further, the community of Xylosandrus crassiusculus may be unique partly, because the species was introduced into the United States only several decades ago (Anderson, 1974). Non-native populations that originate from small initial populations may lose heritable associates during the establishment bottleneck (Rey et al., 2013). The fact that X. crassiusculus is dominated by Ambrosiella sp. is likely a true pattern, as Ambrosiella sp. has been reported as a dominant associate of the beetle in its native East Asia (Kinuura, 1995) and of other species from the same phylogenetic clade (that is, Cnestus; Six et al., 2009). Studies of additional beetle species are needed to test the pattern of correlation between fungal community structure and mycangium type.
The core associates
Fungal OTUs were divided into core species and transient species based on the position of each OTU within the SAD (Figures 3 and 4, Supplementary Tables S5 and S8). OTU sets that followed log-normal distribution were considered core associates. Inconsistent OTUs that fit a log-series distribution of abundances were considered transient. The core community consisted of 8 fungal OTUs (48.2% OTUs, 78% of sequences) for X. affinis and 6 OTUs (42.3% of OTUs, 64% of reads) for X. ferrugineus. In X. crassiusculus, the core consisted of 11 OTUs (62.4% OTUs, 86% of reads), but most of them were of low abundance, as 65% of all reads were represented by one dominant symbiont (Figures 2 and 4).

Separating core and transient community members based on discontinuity in persistence/abundance distribution of fungal OTUs. Dashed lines are segregating OTU sets that fit the log-normal abundance distribution (core symbionts, right) and log-series distribution (transient associates, left).

Distribution of abundance classes of core species (transient, incidental species excluded, see Materials and methods section for details). Red curve shows expected log-normal distribution of abundances. From left to right: X. affinis, X. ferrugineus, X. crassiusculus. Communities in both Xyleborus are more evenly abundant than that in Xylosandrus crassiusculus.
In summary, each beetle species appears to have multiple core associates. However, communities in both Xyleborus spp. have much more even abundances than those in Xylosandrus, where the dominant species (Ambrosiella sp.) is >10 × more abundant than most core species. (Supplementary Table S4). This is consistent with our semi-quantitative reference cultures (Supplementary Tables S6 and S7).
Taxonomic identity of the fungal community
We have recovered genera that have been reported as the dominant mutualistic symbionts, primarily the genus Ambrosiella (Microascales; Table 3, Supplementary Table S8). Identification of this genus was possible only through a comparison with the reference communities included in the sequencing run. Identification directly by BLAST or RDP was impossible due to the weak representation of Ambrosiella spp. in public databases, unsettled nomenclature and the conserved sequence of the ITS2 marker, as discussed above.
For example, the OTU HQ538467, which was abundant and frequent in both Xyleborus sp., did not have a match in taxonomic databases. It is most likely Raffaelea sp. as it matched our Raffaelea-only reference community. The Raffaelea-like OTU was rare in X. crassiusculus, but an order of magnitude more common in both Xyleborus species. The genus Raffaelea appears to be the primary symbiont of Xyleborus spp. based on literature (Harrington et al., 2010) and on our reference cultures (Supplementary Tables S6 and S7). Its closest match in taxonomic databases was an undetermined Ceratocystis sp. Undersampling of Raffaeleas spp. and other ophiostomatoid associates in our data set was likely due to amplification inefficiency and template competition in complex fungal communities. ITS sequences of these taxa are also rare in public databases, further illustrating the problematic amplification in such taxa (for example, Fraedrich et al., 2008).
In addition to the known mutualists, we also detected many other fungi that are sufficiently frequent to qualify for the statistical core community but that have never been reported from the ambrosia symbiosis (Table 1, Supplementary Table S4). The ecological function of these fungi and their role in the ambrosia consortium cannot be inferred from sequence data. However, the genera to which these fungi belong do have well-characterized ecologies, which allows us to predict potential function.
-
One of the most frequently detected and well-identified species was Diplodia corticola—a tree pathogen that has recently invaded the United States and is increasingly damaging to economically important trees (Úrbez-Torres et al., 2010; Dreaden et al., 2011; Lynch et al., 2013). This indicates that not all fungi transported by ambrosia beetles are involved in the symbiosis and that ambrosia beetles may frequently transport tree pathogens.
-
Another example of a high-frequency associate with unknown functions was Fusarium. The ITS marker is not suitable for identification of fusaria to the species level (O'Donnell, 2000; Balajee et al., 2009). Fusaria are frequently recovered from wood-boring beetles as common phoretic contaminants (Batra, 1963, 1967). However, several clades in the Fusarium solani complex are also intimately associated with at least three clades of ambrosia beetles (Norris, 1979; Kasson et al., 2013). We do not know whether the common Fusarium OTU detected within the mycangia of beetles investigated here was symbiotic or phoretic and whether it is one species or several.
-
Numerous OTUs represented different lineages of yeasts or yeast-like fungi, both ascomycetous (Saccharomycotina) and basidiomycetous. An example is an OTU identified as either Cryptococcus sp. or Bandoniozyma glucofermentas, which was frequent in all three beetle species, and most abundant in the more permissive mandibular mycangia of Xyleborus. Yeasts are common associates of many wood-boring beetles (Ganter, 2006; Rivera et al., 2009), not only ambrosia beetles, and appear to be specialized to the habitat rather than to the vector (Calderon and Berkov, 2012). Yeast may compete with beetles for some of the same nutrients (that is, glucose in B. glucofermentas (Valente et al., 2012)), but they may also be a source of vitamins and other essential metabolites.
Many other fungi were present whose genera, identified by BLAST, suggest diverse life strategies not related to beetles. These include plant pathogens, epiphytes, endophytes, soil saprophytes and common molds, such as Cladosporium sp.
Culture-independent assessments of community structure are becoming essential for describing symbioses, be it the ambrosia beetle–fungus system or other fungus-farming systems or any other multi-member symbiotic complex. However, quantities of microbial members are only an approximation of their importance for the macrosymbiont. In our case, many of the taxa that dominated the sequencing output were indeed the coevolved nutritional symbionts, but we know that only because of external experimental evidence. Other taxa in our output were equally abundant, yet we cannot conclude whether that is due to their ecological significance or due to incidental causes, such as prolific spore-forming or amplification bias. We caution against overinterpreting next-generation sequencing data as a reflection of ecological functions in symbiology.
Factors that shape the fungal community composition
Beetle species was a much stronger predictor of fungus community composition than locality (Figure 5). This is suggested by both nonparametric tests (ANOSIM and PERMANOVA, Table 4), as well as by the Principal Coordinate Analysis, in which samples labeled by beetle species show distinct clustering, whereas samples labeled by location do not (Figure 5). However, the effect of locality may have been reduced by the limited geographic range of our samples, which is in contrast with the nearly global distribution of all three beetle species. There was also a significant influence of the interaction between beetle species and locality; however, its implications are unclear, and it may be a result of uneven sampling.

Principal Coordinate Analysis plots of fungal communities in mycangia of Xyleborus affinis, X. ferrugineus and Xylosandrus crassiusculus based on Bray–Curtis dissimilarities. (a) Samples labeled according to the ambrosia beetle species of origin. (b) Samples labeled according to the geographic location of origin.
The differences between fungal communities in the two beetle genera may be mediated by the different types of mycangia. Several groups of fungi, whose function in the beetle is unknown, were significantly more abundant in the mandibular mycangia of the two Xyleborus species than in the mesonotal mycangium of Xylosandrus. Xylosandrus contained a greater number of species in total, including numerous transient fungi, but the majority of them represented only fractions of abundance. This pattern supports the hypothesis that the mandibular mycangium, although much smaller and concealed, provides a more permissive environment for a broader array of symbionts, as well as, nonspecific fungi. The thoracic mycangium of Xylosandrus also collects nonspecific fungi, possibly as spores, but appears to better restrict their proliferation.
Statistical differences between fungal communities were driven by many fungal species, more than just by the known primary symbionts (Supplementary Table S8). As many as 10 different OTUs were significantly associated with only 1 or 2 beetle species (ANOVA and G-test; Supplementary Tables S4 and S8). These are statistical associations, and their ecological relevance remains to be studied.
Conclusions
Ambrosia beetle mycangia contain one or, in some beetle species, a few of the most coevolved nutritional symbionts, but the fungal community as a whole is more diverse and more promiscuous than previously documented. The promiscuity appears to differ significantly between beetle lineages. Each lineage, equipped with a distinct organ for fungus transmission, carries an overlapping but different set of associates and contaminants, each with their own degree of fidelity and community structure. Horizontal exchange of fungi may be common (Carillo et al., 2013), but each mycangium type supports the assembly of a distinct community.
Our results also corroborate some of the beetle–fungus associations reported in the literature (the second hypothesis, see Table 1). However, publications from the premolecular era vary significantly in identification accuracy and report only the most common and easily culturable fungi, which represent only a fraction of the actual diversity discovered here.
To avoid further spread of the uncertainty about beetle–fungus associations that has plagued the ambrosia symbiosis literature, we recommend that future surveys of this unusually diverse system include careful extraction techniques, sufficient sample sizes, phylogeny-informed species selection and a combination of culture-dependent and independent methods.
Accession codes
References
Abarenkov K, Henrik Nilsson R, Larsson KH, Alexander IJ, Eberhardt U, Erland S et al. (2010). The UNITE database for molecular identification of fungi—recent updates and future perspectives. New Phytol 186: 281–285.
Amend AS, Seifert KA, Bruns TD . (2010). Quantifying microbial communities with 454 pyrosequencing: does read abundance count? Mol Ecol 19: 5555–5565.
Anderson D . (1974). First record of Xyleborus semiopacus in the continental United States (Coleoptera, Scolytidae). Cooperative Economic Insect Report 24: 863–864.
Anderson MJ . (2001). A new method for non-parametric multivariate analysis of variance. Austral Ecol 26: 32–46.
Arfi Y, Buee M, Marchand C, Levasseur A, Record E . (2012). Multiple markers pyrosequencing reveals highly diverse and host-specific fungal communities on the mangrove trees Avicennia marina and Rhizophora stylosa. FEMS Microbiol Ecol 79: 433–444.
Baker JM, Norris DM . (1968). A complex of fungi mutualistically involved in the nutrition of the ambrosia beetle Xyleborus ferrugineus. J Invertebr Pathol 11: 246–250.
Balajee S, Borman A, Brandt M, Cano J, Cuenca-Estrella M, Dannaoui E et al. (2009). Sequence-based identification of Aspergillus, Fusarium, and Mucorales species in the clinical mycology laboratory: where are we and where should we go from here? J Clin Microbiol 47: 877–884.
Batra LR . (1963). Ecology of ambrosia fungi and their dissemination by beetles. Trans Kansas Acad Sci (1903-) 66: 213–236.
Batra LR . (1966). Ambrosia fungi: extent of specificity to ambrosia beetles. Science 153: 193–195.
Batra LR . (1967). Ambrosia fungi - a taxonomic revision and nutritional studies of some species. Mycologia 59: 976–1017.
Beaver RA . (1989). Insect–fungus relationships in the bark and ambrosia beetles. In: Wilding N, Collins NM, Hammond PM, Webber JF (eds). Insect–fungus Interactions. Academic Press: London, UK, pp 121–143.
Bellemain E, Carlsen T, Brochmann C, Coissac E, Taberlet P, Kauserud H . (2010). ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. BMC Microbiol 10: 189.
Benjamini Y, Hochberg Y . (1995). Controlling the false discovery rate - a practical and powerful approach to multiple testing. J Roy Stat Soc B Met 57: 289–300.
Berry D, Ben Mahfoudh K, Wagner M, Loy A . (2011). Barcoded primers used in multiplex amplicon pyrosequencing bias amplification. Appl Environ Microbiol 77: 7846–7849.
Biedermann PH, Klepzig KD, Taborsky M . (2009). Fungus cultivation by ambrosia beetles: behavior and laboratory breeding success in three xyleborine species. Environ Entomol 38: 1096–1105.
Bruns TD, Shefferson RP . (2004). Evolutionary studies of ectomycorrhizal fungi: recent advances and future directions. Can J Bot 82: 1122–1132.
Caldera EJ, Poulsen M, Suen G, Currie CR . (2009). Insect symbioses: a case study of past, present, and future fungus-growing ant research. Environ Entomol 38: 78–92.
Calderon O, Berkov A . (2012). Midgut and fat body bacteriocytes in neotropical cerambycid beetles (Coleoptera: Cerambycidae). Environ Entomol 41: 108–117.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Carrillo D, Duncan RE, Ploetz JN, Campbell AF, Ploetz RC, Peña JE . (2014). Lateral transfer of a phytopathogenic symbiont among native and exotic ambrosia beetles. Plant Pathol 63: 54–62.
Clarke KR . (1993). Non-parametric multivariate analyses of changes in community structure. Austral Ecology 18: 117–143.
Cognato AI, Hulcr J, Dole SA, Jordal BH . (2011). Phylogeny of haplo–diploid, fungus-growing ambrosia beetles (Curculionidae: Scolytinae: Xyleborini) inferred from molecular and morphological data. Zool Scripta 40: 174–186.
Doane RW, Gilliland OJ . (1929). Three California ambrosia-beetles. J Econ Entomol 22: 915–921.
Douglas AE . (2010) The Symbiotic Habit. Princeton University Press: Princeton, NJ, USA.
Dreaden TJ, Shin K, Smith JA . (2011). First report of Diplodia corticola causing branch cankers on live oak (Quercus virginiana) in Florida. Plant Dis 95: 1027–1027.
Edgar RC . (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R . (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27: 2194–2200.
Faircloth BC, Glenn TC . (2012). Not all sequence tags are created equal: designing and validating sequence identification tags robust to indels. PLoS One 7: e42543.
Farrell BD, Sequeira AS, O'Meara BC, Normark BB, Chung JH, Jordal BH . (2001). The evolution of agriculture in beetles (Curculionidae: Scolytinae and Platypodinae). Evolution 55: 2011–2027.
Fraedrich SW, Harrington TC, Rabaglia RJ, Ulyshen MD, Mayfield AE, Hanula JL et al. (2008). A fungal symbiont of the redbay ambrosia beetle causes a lethal wilt in redbay and other Lauraceae in the southeastern United States. Plant Dis 92: 215–224.
Francke-Grosmann H . (1956). Hautdrüsen als träger der pilzsymbiose bei ambrosiakäfern. Z Morph Oekol Tiere 45: 275–308.
Ganter PF . (2006). Yeast and invertebrate associations. In: Péter G, Rosa C (eds). Biodiversity and Ecophysiology of Yeasts. Springer: Berlin, Heidelberg, Germany, pp 303–370.
Gardes M, Bruns TD . (1993). ITS primers with enhanced specificity for basidiomycetes—application to the identification of mycorrhizae and rusts. Mol Ecol 2: 113–118.
Gebhardt H, Begerow D, Oberwinkler F . (2004). Identification of the ambrosia fungus of Xyleborus monographus and X. dryographus (Coleoptera: Curculionidae, Scolytinae). Mycol Progr 3: 95–102.
Gilbert SF, Sapp J, Tauber AI . (2012). A symbiotic view of life: we have never been individuals. Q Rev Biol 87: 325–341.
Harrington TC, Aghayeva DN, Fraedrich SW . (2010). New combinations in Raffaelea, Ambrosiella, and Hyalorhinocladiella, and four new species from the redbay ambrosia beetle, Xyleborus glabratus. Mycotaxon 111: 337–361.
Harrington TC, Fraedrich SW . (2010). Quantification of propagules of the laurel wilt fungus and other mycangial fungi from the redbay ambrosia beetle, Xyleborus glabratus. Phytopathology 100: 1118–1123.
Herrera Isla L, Grillo Ravelo H . (1989). Spathodea campanulata Beauv., new host plant of Ceratocystis fimbriata Hell & Halst and Xyleborus spp. Centro Agrícola 16: 91–93.
Hubbard HG . (1896). Ambrosia beetles. US Dep Agric Yearbook 1896: 421–430.
Hubbard HG . (1897). The ambrosia beetles of the United States. US Dep Agric Entomol Bull 7: 9–30.
Hulcr J, Kolarik M, Kirkendall LR . (2007). A new record of fungus-beetle symbiosis in Scolytodes bark beetles (Scolytinae, Curculionidae, Coleoptera). Symbiosis 43: 151–159.
Hulcr J, Cognato AI . (2010). Repeated evolution of crop theft in fungus-farming ambrosia beetles. Evolution 64: 3205–3212.
Hulcr J, Dunn RR . (2011). The sudden emergence of pathogenicity in insect-fungus symbioses threatens naive forest ecosystems. Proc Biol Sci 278: 2866–2873.
Ihrmark K, Bodeker IT, Cruz-Martinez K, Friberg H, Kubartova A, Schenck J et al. (2012). New primers to amplify the fungal ITS2 region—evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol Ecol 82: 666–677.
Jumpponen A, Jones KL . (2009). Massively parallel 454 sequencing indicates hyperdiverse fungal communities in temperate Quercus macrocarpa phyllosphere. New Phytol 184: 438–448.
Kajimura H, Hijii N . (1992). Dynamics of the fungal symbionts in the gallery system and the mycangia of the ambrosia beetle, Xylosandrus-Mutilatus (Blandford) (Coleoptera, Scolytidae) in relation to its life-history. Ecol Res 7: 107–117.
Kasson MT, O'Donnell K, Rooney AP, Sink S, Ploetz RC, Ploetz JN et al. (2013). An inordinate fondness for Fusarium: phylogenetic diversity of fusaria cultivated by ambrosia beetles in the genus Euwallacea on avocado and other plant hosts. Fungal Genet Biol 56: 147–157.
Kinuura H . (1995). Symbiotic fungi associated with ambrosia beetles. Jpn Agr Res Q 29: 57–63.
Koljalg U, Nilsson RH, Abarenkov K, Tedersoo L, Taylor AF, Bahram M et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol Ecol 22: 5271–5277.
Kunin V, Engelbrektson A, Ochman H, Hugenholtz P . (2010). Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol 12: 118–123.
Kühnholz S . (2004) Chemical ecology and mechanisms of reproductive isolation in ambrosia beetles PhD thesis Simon Fraser University: Burnaby, BC, Canada.
Leach JG, Hodson AC, Chilton SJP, Christensen CM . (1940). Observations on two ambrosia beetles and their associated fungi. Phytopathology 30: 227–236.
Lynch S, Eskalen A, Zambino P, Scott T . (2013). First report of bot canker caused by Diplodia corticola on coast live oak (Quercus agrifolia) in California. Forest Ecol Manage 291: 30–42.
Magurran AE, Henderson PA . (2003). Explaining the excess of rare species in natural species abundance distributions. Nature 422: 714–716.
Massoumi Alamouti S, Tsui CK, Breuil C . (2009). Multigene phylogeny of filamentous ambrosia fungi associated with ambrosia and bark beetles. Mycol Res 113: 822–835.
Nilsson RH, Ryberg M, Abarenkov K, Sjokvist E, Kristiansson E . (2009). The ITS region as a target for characterization of fungal communities using emerging sequencing technologies. FEMS Microbiol Lett 296: 97–101.
Nilsson RH, Veldre V, Hartmann M, Unterseher M, Amend A, Bergsten J et al. (2010). An open source software package for automated extraction of ITS1 and ITS2 from fungal ITS sequences for use in high-throughput community assays and molecular ecology. Fungal Ecol 3: 284–287.
Norris DM . (1979). The mutualistic fungi of Xyleborini beetles. In: Batra LR (ed). Insect–Fungus Symbiosis: Nutrition, Mutualism, and Commensalism. Wiley: New York, NY, USA, pp 53–63.
O'Donnell K . (2000). Molecular phylogeny of the Nectria haematococca-Fusarium solani species complex. Mycologia 92: 919–938.
Ploetz R, Harrington T, Hulcr J, Fraedrich S, Smith J, Inch S et al. (2011). Recovery Plan for Laurel Wilt of Avocado (caused by Raffaelea lauricola). National Plant Disease Recovery System. Homeland Security Presidential Directive Number 9 (HSPD-9).
Quince C, Lanzen A, Curtis TP, Davenport RJ, Hall N, Head IM et al. (2009). Accurate determination of microbial diversity from 454 pyrosequencing data. Nat Methods 6: 639–641.
R Core Team. (2013) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, URL http://www.R-projectorg.
Reeder J, Knight R . (2010). Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat Methods 7: 668–669.
Rey O, Estoup A, Facon B, Loiseau A, Aebi A, Duron O et al. (2013). Distribution of endosymbiotic reproductive manipulators reflects invasion process and not reproductive system polymorphism in the little fire ant Wasmannia auropunctata. PLoS One 8: e58467.
Rivera FN, Gonzalez E, Gomez Z, Lopez N, Hernández-Rodríguez C, Berkov A et al. (2009). Gut-associated yeast in bark beetles of the genus Dendroctonus Erichson (Coleoptera: Curculionidae: Scolytinae). Biol J Linnean Soc 98: 325–342.
Roeper R . (1996). Ambrosia beetles of the continental United States and Canada and status of knowledge of their associated primary symbiotic fungi. Mich Entomol Soc Newsl 41: 12–14.
Saunders JL, Knoke JK . (1967a). Diurnal emergence of Xyleborus ferrugineus (Coleoptera - Scolytidae) from cacao trunks in Ecuador and Costa Rica. Ann Entomol Soc Am 60: 1094–1096.
Saunders JL, Knoke JK . (1967b). Diets for rearing the ambrosia beetle Xyleborus ferrugineus (Fabricius) in vitro. Science 157: 460–463.
Schmidt PA, Balint M, Greshake B, Bandow C, Rombke J, Schmitt I . (2013). Illumina metabarcoding of a soil fungal community. Soil Biol Biochem 65: 128–132.
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA et al. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci USA 109: 6241–6246.
Six DL, Stone WD, de Beer ZW, Woolfolk SW . (2009). Ambrosiella beaveri, sp. nov., associated with an exotic ambrosia beetle, Xylosandrus mutilatus (Coleoptera: Curculionidae, Scolytinae), in Mississippi, USA. Antonie Van Leeuwenhoek 96: 17–29.
Unterseher M, Jumpponen A, Opik M, Tedersoo L, Moora M, Dormann CF et al. (2011). Species abundance distributions and richness estimations in fungal metagenomics—lessons learned from community ecology. Mol Ecol 20: 275–285.
Úrbez-Torres J, Peduto F, Rooney-Latham S, Gubler W . (2010). First report of Diplodia corticola causing grapevine (Vitis vinifera) cankers and trunk cankers and dieback of canyon live oak (Quercus chrysolepis) in California. Plant Dis 94: 785–785.
Valente P, Boekhout T, Landell MF, Crestani J, Pagnocca FC, Sette LD et al. (2012). Bandoniozyma gen. nov., a genus of fermentative and non-fermentative tremellaceous yeast species. PLoS One 7: e46060.
Van der Walt JP . (1972). The yeast genus Ambrosiozyma gen. nov.(Ascomycetes). Mycopathol Mycol Appl 46: 305–315.
Verrall AF . (1943). Fungi associated with certain ambrosia beetles. J Agric Res 66: 0135–0144.
Wang Q, Garrity GM, Tiedje JM, Cole JR . (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267.
Weber BC, McPherson JE . (1984). The ambrosia fungus of Xylosandrus-Germanus (Coleoptera, Scolytidae). Can Entomol 116: 281–283.
White TJ, Bruns T, Lee S, Taylor J . (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protocols: A Guide to Methods and Applications 18: 315–322.
Wood SL, Bright DE . (1992). A Catalog of Scolytidae and Platypodidae (Coleoptera). Part 2: Taxonomic Index, Volumes A and B. Great Basin Naturalist Memoirs 13.
Zimmermann G, Butin H . (1973). Untersuchungen über die Hitze-und Trockenresistenz holzbewohneneder Pilze. Flora 162: 393–419.
Acknowledgements
The project was funded by the USDA Forest Service cooperative agreements 12-CA-11420004-042 and 11-CA-11330129-092, the National Science Foundation grant DEB 1256968, the NIFA grant 2009-51181-05915, grant 214232/F20 from the Norwegian Research Council and in-kind contributions. We thank Rob Dunn for support in the methods-development phase and Paul Kendra and Chris Gibbard for the supply of specimens.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Kostovcik, M., Bateman, C., Kolarik, M. et al. The ambrosia symbiosis is specific in some species and promiscuous in others: evidence from community pyrosequencing. ISME J 9, 126–138 (2015). https://doi.org/10.1038/ismej.2014.115
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2014.115
This article is cited by
-
Fungi associated with the ambrosia beetle Xyleborus perforans (Coleoptera: Curculionidae: Scolytinae) on drought-stressed Pinus in New South Wales, Australia
Australasian Plant Pathology (2024)
-
Microorganisms Associated with the Ambrosial Beetle Xyleborus affinis with Plant Growth-Promotion Activity in Arabidopsis Seedlings and Antifungal Activity Against Phytopathogenic Fungus Fusarium sp. INECOL_BM-06
Microbial Ecology (2023)
-
Fungal symbiont community and absence of detectable mycangia in invasive Euplatypus ambrosia beetles
Symbiosis (2023)
-
Two Apriona Species Sharing a Host Niche Have Different Gut Microbiome Diversity
Microbial Ecology (2022)
-
The gut microbiome analysis of Anastrepha obliqua reveals inter-kingdom diversity: bacteria, fungi, and archaea
Archives of Microbiology (2022)
