
Abstract
The Amazon basin harbors a diverse ecological community that has a critical role in the maintenance of the biosphere. Although plant and animal communities have received much attention, basic information is lacking for fungal or prokaryotic communities. This is despite the fact that recent ecological studies have suggested a prominent role for interactions with soil fungi in structuring the diversity and abundance of tropical rainforest trees. In this study, we characterize soil fungal communities across three major tropical forest types in the western Amazon basin (terra firme, seasonally flooded and white sand) using 454 pyrosequencing. Using these data, we examine the relationship between fungal diversity and tree species richness, and between fungal community composition and tree species composition, soil environment and spatial proximity. We find that the fungal community in these ecosystems is diverse, with high degrees of spatial variability related to forest type. We also find strong correlations between α- and β-diversity of soil fungi and trees. Both fungal and plant community β-diversity were also correlated with differences in environmental conditions. The correlation between plant and fungal richness was stronger in fungal lineages known for biotrophic strategies (for example, pathogens, mycorrhizas) compared with a lineage known primarily for saprotrophy (yeasts), suggesting that this coupling is, at least in part, due to direct plant–fungal interactions. These data provide a much-needed look at an understudied dimension of the biota in an important ecosystem and supports the hypothesis that fungal communities are involved in the regulation of tropical tree diversity.
Similar content being viewed by others


Introduction
Tropical rainforests are among the most species-rich communities on the planet. Many groups of organisms reach peaks of richness and biomass in the tropics (Gaston, 2000) and much ecological research has been dedicated to the causes and consequences of this diversity (Connell, 1978; Wright, 2002). Understanding the ecological dynamics of these ecosystems is critical because they have a major role in the shaping the Earth’s biosphere and are currently under great pressure from anthropogenic activities (Gullison et al., 2007). Although a rich literature has investigated the ecology of plant and animal communities in the tropics, very little is known about the communities of organisms traditionally studied by microbiologists, such as bacteria and fungi, in tropical systems. This is a major oversight given the growing recognition that the microbiome has a key role in mediating ecological (Friesen et al., 2011) and geochemical processes (Falkowski et al., 2008). Additionally, recent field studies have suggested a prominent role for direct interactions with soil organisms, and in particular fungi, in structuring the diversity and abundance of tropical rainforest trees (Augspurger, 1983; Comita et al., 2010; Mangan et al., 2010; Liu et al., 2012).
Despite the critical role hypothesized for soil fungi in driving tropical rainforest community structure, most plant ecological studies have treated the soil fungal community as a black box. Basic questions remain unanswered about the relative diversity of fungi in tropical systems (Arnold and Lutzoni, 2007; Amend et al., 2010; Tedersoo et al., 2012), their degree of spatial turnover both within and among habitat types (Peay et al., 2010; McGuire et al., 2012) and the strength of linkage with plant communities (May, 1991; Gilbert and Sousa, 2002; Gilbert et al., 2002). Knowing the details of fungal community structure has important implications for the development of ecological theory in tropical forests. For example, negative feedbacks are generally thought to depend on high levels of variability in the strength of plant–fungal interactions and on strong spatial heterogeneity in fungal community composition (Gilbert, 2002). However, theoretical (May, 1991) and empirical (Gilbert et al., 2002; Smith et al., 2011, 2013) studies have suggested low levels of fungal host specificity in tropical forests, and many studies have questioned the importance of dispersal limitation in microbes (Finlay, 2002; Queloz et al., 2011). Detailed knowledge on the structure of fungal communities is thus necessary to guide research to answer these critical ecological questions.
In this study, we characterize fungal community structure across large spatial and environmental gradients in the Peruvian Amazon basin using next-generation DNA sequencing. We use these data to (a) characterize local-scale fungal richness (α-diversity) and test whether fungal richness is positively correlated with local plant species richness; (b) quantify spatial turnover (β-diversity) in fungal community similarity and test whether or not this is correlated with plant community structure. Few previous studies have looked at fungal community structure in great depth or across large gradients in tropical forests. Our results show that fungal diversity is high in tropical soils and that there is a strong linkage between plant and fungal communities. We discuss how these results can shed light on the potential for plant–soil feedbacks to regulate tropical tree community structure.
Materials and methods
Sites
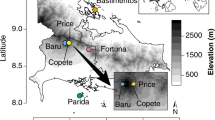
All sites were located in the western Amazon basin in Loreto, Peru, as part of a larger ongoing project examining the effects of soil heterogeneity on tropical forest structure and floristic composition (Fine et al., 2010; Baraloto et al., 2011). The region is aseasonal and receives an annual average of >3000 mm of rain (Marengo, 1998). The sites sampled were chosen to be representative of undisturbed tropical rainforest and have no known history of logging, fire or other disturbance.
We sampled fungal communities in seven plots that represent strong natural gradients in tree species diversity and community structure, with two plots each in seasonally flooded and white sand forests and three plots in terra firme clay forests. Plots were established in July 2008 following methods for modified ‘Gentry plots’ as described in Phillips et al., 2003, with 10 aggregate 2 × 50 m transects (0.1 ha) within a 2-ha area. In our plots, transects are arranged along a central line (plot center) in a ‘wishbone’ format (Supplementary Figure S1). In all the plots, we recorded (i) soil characteristics and forest structure; and (ii) species identity for all the trees >2.5 cm diameter at 1.3 m height, standardizing taxonomy across regions with project-specific vouchers referenced to the Missouri Botanic Garden’s herbarium (see Supplementary Table S1 for complete plant species list). All environmental and tree community variables were recorded at the plot-level (for example, one measurement per plot) for the purpose of our statistical analyses. More details of plot design, inventory and measurement of environmental parameters are available in Baraloto et al. (2011). Although we did not measure soil pH at these sites, other studies from the same region suggest that pH can co-vary with other soil environmental variables, such as base cation concentration, that were measured in our plots (Tuomisto et al., 2003, Paul VA Fine, unpublished data).
Fungal community sampling
We took 10 soil samples at 20 m intervals along the center of seven plots (N of 7 × 10=70 samples). Each sample consisted of a 1000 cm3 (10 × 10 × 10 cm) cube removed using a sharp knife. Soil samples were field sieved to remove roots and large debris and then stored in a plastic bag for transportation. On the same day, a single 0.5 g subsample taken from each core were weighed out on a digital microbalance and stored individually in Lifeguard DNA preservation solution (MoBIO, Carlsbad, CA, USA). DNA extraction and analyses were performed on separate samples from soil chemistry. Due to the remote location of most field sites, samples were stored in a cool location but were not refrigerated until return to Iquitos, generally <1 week. In Iquitos, samples were stored at 4 °C until shipment back to UC Berkeley where they were kept at −20 °C until DNA extraction.
Molecular methods
DNA was extracted from each sample using the PowerSoil DNA Isolation Kit (MoBio). For each DNA extraction, we performed a single PCR amplification in a 20-μl reaction using 1 unit of HotStarTaq polymerase (Qiagen, Valencia, CA, USA), 1 × PCR buffer, 200 μM of each dNTP, 0.375 μM of each primer. Cycling parameters were 1 cycle of 95 °C for 3 min, 35 cycles of 94 °C for 1 min, 51 °C for 1 min and 72 °C for 1 min followed by a final extension at 72 °C for 7 min. Forward primer consisted of the 454 Lib-L A-adapter, a 10 base pair barcode, plus the fungal-specific primer ITS-1F (Internal Transcribed Spacer-1F; Gardes and Bruns, 1993) while the reverse primer consisted of 454 Lib-L B-adapter and the ITS-4 primer. Negative controls were included at all stages of the sampling process, including blank Lifeguard samples carried into the field, as well as negative controls introduced during DNA extraction and PCR amplification. Successful amplification was confirmed using gel electrophoresis. Successful amplifications were cleaned using the QIAquick PCR purification kit (Qiagen), quantified using the Quant-iT dsDNA HS Assay Kit on a Qubit fluorometer (Invitrogen, Carlsbad, CA, USA) and pooled into equimolar concentrations. The pooled sample was sequenced using Titanium chemistry on ¼ of a 454 sequencing run using the Lib-L amplicon kit at the Duke University Genome Sequencing and Analysis Core Facility.
Sequence processing
Initial sequence processing and sample assignment were done using the QIIME v.1.3.1 software package (Caporaso et al., 2010) with the flags −l 300 −L 700 −e 0 and −H 10. To account for sequencing error that can arise as part of the PCR and 454 sequencing process, we used flowgram clustering (Knight and Reeder, 2010) as implemented in the QIIME v.1.3.1 software package with a Titanium error profile. Our sequences were clustered into operational taxonomic units (OTUs) at 97% sequence similarity using the USEARCH algorithm (Edgar, 2010), a level that has been shown to approximate known species concepts in fungi (Smith et al., 2007). Chimeric sequences were removed using a combination of de novo and reference-based Chimera checking using the program UCHIME (Edgar et al., 2011) implemented during the USEARCH clustering process. Based on these methods, we observed 108 and 144 sequences with de novo and reference-based Chimera checking, respectively, of which 76 were flagged by both methods (approximately 4% of OTUs) and removed from all further analyses. Taxonomic affiliation of OTUs was assigned for a representative sequence from each OTU using the BLAST option in the assign_taxonomy.py script. Our database consisted of a previously published sequence database (Tedersoo et al., 2010b) and a QIIME-formatted taxonomy file that was initially based on downloaded GenBank taxonomy using a custom PERL script written by Jason Stajich (http://bit.ly/RlzNY5). This file was then manually edited so that all sequences included the same subset of ranks (Kingdom, Phylum, Class, Order, Family and Genus). Phylogenetically uniformative designations (for example, Mitosporic Ascomycetes) were coded as Unknown. Because of known problems with possible skewed sequence abundance using fungal ITS primers (Amend et al., 2010b, Ihrmark et al., 2012) we use only sample presence-absence from our rarefied OTU tables to describe the fungal community.
Statistical analysis
The relationship between per-sample fungal OTU richness and plot variables was analyzed using a linear mixed-effects model with samples nested within plots and plot identity as a random variable in the R programming environment (R Core Development Team, 2009) with the package nlme. Because samples differed in the number of sequence reads obtained (mean=1670, range=0–6071), each sample was rarefied to 500 sequences using the alpha_rarefaction.py script in QIIME. This sequence depth allowed us to retain the majority of samples (54/66) while still maintaining a reasonable sequencing depth. Preliminary analyses using a cutoff of 1000 sequences showed no apparent differences. To see whether the scale of fungal community analysis mattered (for example, sample vs plot level), we calculated total plot-level fungal richness (rarefied to 6500 sequences, the minimum number of sequences across all plots) and used regular linear regression to test for a relationship between plant and fungal richness. Because mean richness per sample and per plot were highly correlated (r2=0.98, P<0.001), the scale of analysis does not affect our results. To evaluate whether the relationship with plant species richness differed across fungal trophic strategies, we also calculated plot-level richness for three of the most diverse and abundant lineages found in the study (Hypocreales, Glomerales, Saccharomycetales), and which have relatively distinct trophic strategies.
Community structure was analyzed by rarefying soil samples to 500 sequences using the multiple_rarefactions.py script in QIIME and calculating community dissimilarity using either the Jaccard or βSim index (Koleff et al., 2003). This process was repeated 10 times and the average dissimilarity score across samples was used for analyses. We chose to use two methods to measure community dissimilarity. Jaccard distance is calculated as 1−A/(A+B+C), where ‘A’=the species found in both the samples, ‘B’=species unique to Sample 1 and ‘C’=species unique to Sample 2. Jaccard distance thus provides a metric based on absolute number of species shared between two samples, ranging from 0 (no species shared) to 1 (all species shared). The βSim metric controls for differences in sample richness by using the minimum number of species, which could be shared to calculate disimilarity, for example, min(B,C)/(min(B,C)+A). Because the two methods gave similar results, we report results based on Jaccard here due to its ease of interpretation. A Mantel test was used to determine whether or not samples taken from a given forest were independent or showed spatial autocorrelation. To test whether habitat influenced community structure, we calculated the proportion of sample variation attributable to habitat type using permutational analysis of variance (Anderson, 2001) as implemented by the function Adonis in the R package Vegan. Plot-level fungal β-diversity was also calculated and regressed against plant β-diversity, spatial distance between plots and environmental similarity, using the Mantel test in Vegan. Because most environmental variables are correlated, environmental similarity was calculated as Euclidean distance calculated from scaled principal component analysis scores for each site based on 12 soil variables, including total C, N, C:N ratio, NO3-N, P, K, Na, Ca, Mg, % Sand, % Silt and % Clay. For details, see Baraloto et al. (2011).
Results
Fungal α-diversity varied greatly across our samples but tended to mirror patterns of tree α-diversity. In total, we detected 1776 species-level fungal OTUs from our successfully sequenced soil samples. Average fungal richness per soil sample (rarefied to 500 sequences) was similar in magnitude (mean=44, s.d.=44, range=2–177) to plot-level diversity observed for tree species (mean=111, s.d.=38, range=64–152), while mean plot-level fungal richness (rarefied to 6500 sequences) was 297 (s.d.=222, range=68–629). Chao 1 estimates of richness were about 30% higher on average, with per sample mean of 62 OTUs (s.d.=68, range=2–275) and per plot mean of 389 (s.d.=288, range=97–823). Observed fungal OTU richness was positively correlated with tree species richness at both the sample (slope=0.83, t=5.04, P=0.004; Figure 1a) and plot level (r2=0.79, slope=5.12, t=4.31, P=0.008; Figure 1b). Soil environmental variables, on the other hand, showed no significant relationship with fungal OTU richness. Models showed no relationship between fungal sample richness and total soil nitrogen (Slope=−72.43, t=−0.53, P=0.62), soil NO3 (slope=2.47, t=0.97, P=0.38), phosphorus (slope=−4.24, t=−1.92, P=0.08), percentage of soil clay content (slope=−0.19, t=−0.31, P=0.77) or soil C:N ratio (slope=−2.31, t=−1.14, P=0.30). There were also no significant relationships between tree species richness and the same soil environmental variables (data not shown).

Fungal and tree species richness are positively correlated. Figure shows regression of tree species richness per plot against: (a) fungal richness per sample, and (b) fungal richness per plot. For sample-level analyses, fungal richness was treated as a nested variable using a linear mixed-effects model. Both regressions were significant at P<0.05 level.
Fungal β-diversity, or species turnover, was high across both the samples and plots. Mean Jaccard distance, where 0=identical OTUs and 1=no common OTUs, was 0.96 comparing across all the soil samples (range=0.63–1.0, s.d.=0.05) and 0.92 comparing across all the plots (range=0.85–0.98, s.d.=0.04). Although samples taken within a site tended to be more similar to each other (Adonis F6,46=3.11, P=0.001, R2=0.29), we detected little evidence of spatial autocorrelation comparing soil cores within plots as the Mantel test was insignificant for six of seven plots (P>0.05, except Site 13; Mantel r=0.49, P=0.003). Overall most samples within a plot shared few OTUs (mean within-site Jaccard=0.91, range=0.88–0.94, s.d.=0.024). However, community composition was generally more similar in samples taken from the same edaphic habitat (Adonis F2,52=1.95, P<0.001, R2=0.07, Figure 2).

Non-metric multidimensional scaling (NMDS) plot depicting similarity of fungal communities sequenced from individual soil samples taken across three western Amazon basin forest types: seasonally flooded (grey), terre firme (black), and white sand (white). Different symbols within a given forest type show the plot from which samples originated. Despite the high variability in fungal community structure, clustering of samples by forest type (P>0.05) indicates that these edaphically structured plant communities also harbor distinct fungal communities.
Despite the overall low resemblance of both the plant (mean Jaccard=0.96, range=0.81–0.99, sd=0.04) and fungal communities across plots, there was a positive correlation between plant and fungal β-diversity (Mantel r=0.56, P=0.007; Figure 3). A single outlier in this relationship was observed for two plots on white-sand substrates, which tend to share dominant tree species with good dispersal abilities, resulting in lower plant species turnover compared with pairwise comparisons between other habitat types (Fine and Kembel, 2011). Fungal community turnover remained relatively high between these two plots, which are separated by >100 km. There was no correlation between fungal β-diversity and spatial proximity of plots (Mantel r=0.04, P=0.31); however, both fungal and plant community β-diversity were correlated with differences in environmental conditions (Fungal Mantel r=0.46, P=0.02; Plant Mantel r=0.67, P=0.002).

Coupling of plant and fungal β-diversity across western Amazon basin forest sites. The plot shows pairwise community distance (1−Jaccard index) for fungal and plant communities, where 0=identical communities and 1=no shared OTUs. Overall β-diversity was high, with few taxa in either guild shared across sites. However, the positive correlation (Mantel r=0.56, P=0.007) shows that sites with more similar plant communities also had more similar fungal communities, indicative of biotic coupling.
The evolutionary diversity of fungi recovered from these soils was also high at higher taxonomic levels. Fungal OTUs were assigned across 81 orders, 189 families and 538 genera. Ascomycota and Basidiomycota were the most frequently encountered, accounting for 64% and 23% of all observations (Supplementary Figure S2). Overall patterns of taxonomic representation were fairly consistent across the different soil types (Figure 4). The Hypocreales (204 OTUs), Agaricales (178), Saccharomycetales (121), Xylariales (101) and Glomerales (94) were the most diverse and commonly observed orders across the study (Supplementary Figure S2). Most higher fungal taxa contain mixed ecological strategies, but the Hypocreales are well known to contain a large number of plant-associated pathogens and leaf endophytes (Berbee, 2001; Arnold and Lutzoni, 2007), the Glomerales are exclusively involved in arbuscular mycorrhizal symbiosis (Smith and Read, 2008) and the Saccharomycetales (the true yeasts) are primarily saprotrophic (Suh et al., 2006). We observed a positive correlation between tree species richness and richness of Hypocreales (r2=0.68, slope=0.54±0.16, P=0.022) and Glomerales (r2=0.56, slope=0.32±0.1, P=0.051) but not of Saccharomycetales (r2=0.03, slope=−0.03±0.15, P=0.71) (Figure 5).

Taxonomic composition of fungal communities recovered from rainforest soils in the western Amazon basin. Bars show the frequency with which different taxonomic groups were recovered based on 454 pyrosquencing of the fungal ITS region. Lineages with fewer than five occurrences across all the samples were removed from the figure for clarity. This includes the Atractiellomycetes, Blastocladiomycetes, Cystobasidiomycetes, Dacrymycetes, Exobasidiomycetes, Monoblepharidomycetes, Pucciniomycetes and Zoopagomycotina. The results are shown for all the samples and summarized by soil type. In total, >1700 fungal OTUs from 81 orders, 189 families and 538 genera were detected in our data set. Despite differences in community structure across soil types, phylogenetic representation appears largely similar.

Correlation between plant and fungal richness for lineages with different trophic strategies. We regressed plot-level fungal richness against plant richness for three of the most diverse lineages identified in this study and that are characterized by different trophic strategies: the Hypocreales (plant-associated biotrophs, S=204, Rank=1), Saccharomycetales (Saprotrophs, S=121, Rank=3), and Glomerales (Mycorrhizal, S=94, Rank=5). Our results show that there is a strong correlation between plant and fungal richness for the lineages with strong plant associations (P<0.05, black lines) but not saprotrophic lineages (grey line, P>0.05).
Discussion
Our results show that plant and fungal community structure are strongly correlated in one of the most diverse ecosystems on the planet, the rainforests of the western Amazon basin. This finding is particularly notable given the fact that we were working across multiple habitat types and at sites separated by distances ranging from 3 to >100 km and because of the detection difficulties associated with assessing fungal community structure (Peay et al., 2008). The relationship between plant and fungal community structure is of particular interest given the recent findings that plant interactions with the soil community have a strong role in structuring the diversity and abundance of tropical trees (Comita et al., 2010; Mangan et al., 2010; Terborgh, 2012). We found that tree and fungal diversity were strongly correlated at both the sample and plot level. Although it was not possible to include all plants (for example, herbs and lianas) in our sampling, this does suggest a relatively robust correlation. From a theoretical perspective, there is good reason to expect strong links between plant and fungal richness, either due to host specificity of fungal biotrophs (Gilbert and Webb, 2007) or increased resource diversity for saprotrophs (Waldrop et al., 2006). Although we found no significant abiotic predictors of fungal richness, other studies have found that fungal richness responds more strongly to abiotic factors or productivity rather than to plant species richness per se (Waldrop et al., 2006; McGuire et al., 2012). It is unclear why these studies might differ in their results, but to date, there are still few studies of large-scale plant–fungal richness using next generation sequencing techniques. One possibility is that plant–fungal richness relationships vary with scale or abiotic context. Another is that these relationships depend on the genetic marker or method used to assay fungal community structure (ITS+454 in this study, 18S+454 in McGuire et al., 2012, ITS+ribosomal intergenic spacer analysis (RISA) in Waldrop et al., 2006). It is also important to note that our estimates of fungal diversity are not absolute—for example, they would change with greater sequencing depth or with greater number of soil samples. A solid understanding of fungal–plant diversity relationships is critical for a number of ecological applications, for example, predicting global fungal biodiversity (Hawksworth, 1991, 2012; May, 1991). Hopefully with more large-scale studies from the tropics, and using a common sequencing approach, a clearer answer will emerge.
Fungal communities are highly variable in space and time, even within a single forest stand (Izzo et al., 2005). Despite this, we found that there were correlations between the similarity of plant species and similarity of soil fungal OTUs across our plots. Because the differences in forest type across these western Amazon basin sites are driven by well-known edaphic factors (for example, seasonally flooded, terre firme or white sand soils; Baraloto et al., 2011; Fine and Kembel, 2011), it is not possible to determine from an observational study such as ours whether fungal communities were responding primarily to the plant community, the common soil environment or both. Although a number of studies have shown clear relationships between edaphic variables and fungal community structure (reviewed in Peay et al., 2008), experimental studies of host-dependent soil fungi have also shown that even extreme edaphic conditions (for example, serpentine soils) do not pose much of a direct physiological challenge to soil fungi (Branco, 2010) and many common fungi can grow well in extreme environments, for example, the surface of the Chernobyl nuclear reactor (Dadachova and Casadevall, 2008). Landscape-scale studies of temperate Agaricomycetes across a similar climatic and edaphic background have shown that tree community structure has strong effects on fungal communities (Edwards and Zak, 2010). Similarly, at another neotropical rainforest site, Henkel et al. (2002) found that ectomycorrhizal trees and fungi were not restricted by local variation in soil type. If the edaphic variation exhibited by our sites is not likely to be a direct physiological barrier to most fungi, then it seems plausible that the correlation between plant and fungal community structure we observe is driven, at least in part, by the direct effects of plants on fungal community structure. This could be driven by resource specialization by saprotrophs, particularly given the importance of secondary chemicals in tropical plant leaves or by host specificity of biotrophic pathogens or mutualists. The coupling of plant and fungal community structure through direct interactions is plausible given the strong ecological effects that have been demonstrated by fungi on plant communities (Hansen and Goheen, 2000; Bever, 2002; Klironomos, 2002; Arnold et al., 2003). However, whether or not these local interactions create strong links between plant and fungal communities at larger scales has received less study (Feinstein and Blackwood, 2012; McGuire et al., 2012). Our study suggests that local-scale ecological processes (for example, host specificity, habitat specificity) are at least strong enough to create some predictability in the suites of interacting plants and fungi observed across western Amazonian rainforests, despite the high diversity and spatial variability of the organisms in these communities.
Our findings regarding fungal community structure in this system may have implications for natural-enemy-mediated coexistence theories of rainforest tree species diversity. Despite the promise of coexistence theories involving natural enemies (Gillett, 1962; Janzen, 1970; Connell, 1971), previous studies have not investigated the community structure of the organisms purported to provide the mechanism underlying negative density-dependence (Harms et al., 2000; Comita et al., 2010; Mangan et al., 2010; Liu et al., 2012). Even though much of the original work on density-dependent attack by natural enemies focused on insect herbivores (Janzen, 1970; Connell, 1971), a growing body of empirical work has highlighted the important feedbacks between plants and soil-borne fungal pathogens and mycorrhizal fungi (Augspurger, 1983; van der Heijden et al., 1998; Bever, 2002; Klironomos, 2002). This is perhaps not surprising given the potential virulence and host specificity (Gilbert and Webb, 2007) of fungal pathogens as well as their long evolutionary history with plants. However, data on soil fungal communities have been lacking for the tropics, until very recently. Our study provides, at the least, some much needed molecular-based evidence that soil fungal communities show sufficient raw diversity and spatial turnover to be a powerful mechanism underlying natural-enemy-based coexistence of tropical rainforest tree species (Gilbert, 2002).
Although our study does not directly test Janzen–Connell dynamics, it does provide a first-level test of some critical predictions based on the Janzen–Connell hypothesis. First, we showed that local fungal diversity tracks plant diversity across the forests we studied (Figure 1). This result is consistent with some degree of host specificity in fungal communities. Our data also supported the Janzen–Connell-based predictions that fungal β-diversity should be high and should be linked to plant β-diversity. Patterns of spatial turnover in soil microbes are a topic of great interest in microbial ecology (Green et al., 2004; Horner-Devine et al., 2004), and microbes have long been thought to show high local diversity but low regional or global diversity (Bisby, 1943; Green et al., 2004). These patterns are hypothesized to be driven, at least partly, by the prolific dispersal capabilities of microbial organisms. Recent studies of ectomycorrhizal fungi from the neotropics show similar communities over large geographical areas (Smith et al., 2011, 2013), indicating limited dispersal barriers at the timescales over which these regional communities assemble. Truly unlimited dispersal, however, would negate the distance-dependent mortality necessary for microbial pathogens to regulate plant coexistence under Janzen–Connell dynamics because host-specific microbes would be ubiquitous. However, recent studies have challenged the high dispersal capacity of soil fungi (Peay et al., 2012), such that most diversity is unique to an individual sample (Amend et al., 2010), and similarity between samples generally decays over a few meters (reviewed in Peay et al., 2008). Jaccard distance between two samples represents the proportion of species unique to individual samples. In our study, on average >90% of the fungal species-level OTUs were unique to a given soil sample, even within a single plot. Thus, the soil fungal community encountered by a tree seedling will vary dramatically depending on where it establishes. In addition, despite the fact that few species of trees or fungi were shared across plots in this diverse system, community similarity of plants and fungi was positively correlated. These two findings, namely that the fungal community is spatially variable and is linked to plant species identities, are consistent with the Janzen–Connell requirements for dispersal limitation and host specificity. Our findings, coupled with the growing evidence for fungal dispersal limitation (Galante et al., 2011; Peay et al., 2012) and host specificity (Gilbert and Webb, 2007; Liu et al., 2012), are consistent with the patchy distribution necessary for Janzen–Connell dynamics to be effective in maintaining patterns of high community richness in high-diversity tree communities.
A strong caveat in applying our results directly to Janzen–Connell theories, and in the general utility of next generation sequencing in microbial ecology, is the difficulty in assigning trophic strategies to fungi based solely on DNA sequences. This is due to poor coverage in online genetic repositories (particularly for tropical systems), the paucity of natural history knowledge for most species and the polyphyletic nature of ecological strategies in Fungi (Berbee, 2001). We find a stronger correlation between plant species richness and fungal lineage OTU richness for lineages that are known to contain pathogens (Hypocreales) and mycorrhizal fungi (Glomerales) relative to saprotrophs (Saccharomycetales). Although this is suggestive that biotic interactions structure the community differences we observe across forest types, most of the abundant clades in our study, such as the Hypocreales, Agaricales and Xylariales, include pathogens and saprotrophs (Berbee, 2001) as well as plant endophytes (Arnold and Lutzoni, 2007). In addition, while the Saccharomycetales are primarily saprotrophs, as unicellular fungi they may not represent the same resource-acquisition strategies of filamentous saprotrophs. The utility of molecular data such as ours would increase greatly with better sequence coverage from tropical biomes in online genetic databases. Greater sequence depth coupled with ecological characterization of isolates would allow for more accurate taxonomic classification of environmental sequences and assignment into ecological life history strategies.
Our study is among the first to provide a detailed picture of soil fungal community structure in tropical forests using next-generation sequencing methods. Overall, fungal diversity was high, at around 1700 species-level OTUs using a 97% sequence similarity cutoff. Comparison with diversity in other systems is difficult as there are still relatively few published studies examining soil fungal communities with next-generation sequence data. Working in a temperate forest, Buée et al. (2009) identified approximately 1000 OTUs with a slightly greater sequencing effort (166 000 vs 110 000 sequences). However, they analyzed fewer samples at greater sequence depth and did not use flowgram clustering to reduce pyrosequencing error (Knight and Reeder, 2010). Using the 18S locus combined with 454 pyrosequencing, McGuire et al. (2012) found much greater diversity (over 8000 fungal phylotypes) in another neotropical rainforest that might otherwise have been expected to be similar. The difficulty of such comparisons highlights the need for similar sampling and data processing methodology in order to facilitate comparative analyses. In addition, although molecular data provide a powerful window into the ‘black box’, it should always be interpreted with caution given the biases inherent in any methodology. 454 sequence counts for ITS data have been shown to be non-quantitative (Amend et al., 2010b). Although we restricted our analyses to presence/absence data for this reason, high read abundance of Saccharomycetales in some samples could have been due to PCR or sequencing biases. These kinds of differences could arise due to ITS fragment size variation (the most abundant yeast taxa in our study had ITS1 lengths of only ∼60 bp; see also Toju et al., 2012), DNA:biomass ratios in yeasts relative to filamentous fungi or both. Similarly, Cryptococcus (a Basidiomycete yeast) was highly abundant in the Buée et al. (2009) study. Additionally, although our negative control samples were clean throughout the extraction and PCR process, we detected one species in a single sample that was clearly a lab contaminant (Suillus pungens, a northern hemisphere ectomycorrhizal taxon). Similar results have been reported from other next-generation sequencing studies (Tedersoo et al., 2010) and indicate the ease with which single molecules can be detected using these methods. While all other taxa we examined appeared legitimate, automated detection of non-indigenous sequences is not possible given the large number of taxa and poor coverage of fungi in DNA databases. Although this does not suggest that the majority of 454 data is unreliable, it does suggest that caution should be exercised when interpreting next-generation sequencing results. Finally, while next-generation sequencing data provide a deeper look at fungal diversity relative to previous methods (for example, restriction fragment length polymorphism or cloning and sequencing), 454 sequencing of individual PCR reactions amplified from the same sample may differ in their species composition (Tedersoo et al., 2010). Sequencing a single PCR replicate at a depth of a few 100 sequences (as in this study) may not capture all fungal species in a sample and may exaggerate species turnover across samples. Improved protocols (for example, pooling of multiple PCR replicates) and/or sequencing platforms with deeper sequencing capabilities may resolve these problems in the future. This is one of the few fungal deep-sequencing studies from the Amazon basin and clearly more work is needed, using a variety of molecular approaches and additional sites, in order to substantiate the initial patterns we describe here.
Conclusion
Fungi have a promising role in explaining the relative abundance and coexistence of plant species in terrestrial communities. This is particularly exciting in tropical forests where abiotic niche partitioning alone appears insufficient to explain coexistence (Hubbell, 2001). We show here that fungal community structure is strongly linked with plants, both in terms of species composition and species richness. These results shed important light on an understudied group of organisms in a an important ecosystem. Field experiments to characterize the ecology of fungi from these systems and to disentangle the biotic and abiotic drivers of fungal community structure are critical next steps in elucidating the important role of fungi in structuring tropical ecosystems.
References
Amend A, Samson R, Seifert KA, Bruns TD . (2010). Indoor fungal composition is geographically patterned and more diverse in temperate zones than in the tropics. Proc Natl Aacd Sci USA 107: 13748–13753.
Anderson MJ . (2001). A new method for non-parametric multivariate analysis of variance. Aust Ecol 26: 32–46.
Arnold AE, Lutzoni F . (2007). Diversity and host range of foliar fungal endophytes: are tropical leaves biodiversity hotspots? Ecology 88: 541–549.
Arnold AE, Mejia LC, Kyllo D, Rojas EI, Maynard Z, Robbins N et al (2003). Fungal endophytes limit pathogen damage in a tropical tree. Proc Natl Acad Sci USA 100: 15649–15654.
Augspurger CK . (1983). Seed dispersal of the tropical tree, Platypodium elegans, and the escape of its seedlings from fungal pathogens. J Ecol 71: 759–771.
Baraloto C, Rabaud S, Molto Q, Blanc L, Fortunel C, Hérault B et al (2011). Disentangling stand and environmental correlates of aboveground biomass in Amazonian forests. Global Change Biol 17: 2677–2688.
Berbee ML . (2001). The phylogeny of plant and animal pathogens in the Ascomycota. Physiol Mol Plant Pathol 59: 165–187.
Bever JD . (2002). Negative feedback within a mutualism: host-specific growth of mycorrhizal fungi reduces plant benefit. Proc R Soc B 269: 2595–2601.
Bisby GR . (1943). Geographical distribution of fungi. Bot Rev 9: 466–482.
Branco S . (2010). Serpentine soils promote ectomycorrhizal fungal diversity. Mol Ecol 19: 5566–5576.
Buée M, Reich M, Murat C, Morin E, Nilsson RH, Uroz S et al (2009). 454 Pyrosequencing analyses of forest soils reveals an unexpectedly high fungal diversity. New Phytologist 184: 449–456.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Comita LS, Muller-Landau HC, Aguilar S, Hubbell SP . (2010). Asymmetric density dependence shapes species abundances in a tropical tree community. Science 329: 330–332.
Connell JH . (1971). On the role of natural enemies in preventing competitive exclusion in some marine animals and in rain forest trees. In: Den Boer PJ, Gradwell GR, (eds). Dynamics of Populations. PUDOC: Wageningen, the Netherlands, pp 298–312.
Connell JH . (1978). Diversity in tropical rain forests and coral reefs—high diversity of trees and corals is maintained only in a non-equilibrium state. Science 199: 1302–1310.
Dadachova E, Casadevall A . (2008). Ionizing radiation: how fungi cope, adapt, and exploit with the help of melanin. Curr Opin Microbiol 11: 525–531.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R . (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27: 2194–2200.
Edgar RC . (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461.
Edwards IP, Zak DR . (2010). Phylogenetic similarity and structure of Agaricomycotina communities across a forested landscape. Mol Ecol 19: 1469–1482.
Falkowski PG, Fenchel T, Delong EF . (2008). The microbial engines that drive Earth’s biogeochemical cycles. Science 320: 1034–1039.
Feinstein LM, Blackwood CB . (2012). Taxa-area relationship and neutral dynamics influence the diversity of fungal communities on senesced tree leaves. Environ Microbiol 14: 1488–1499.
Fine PVA, García-Villacorta R, Pitman NCA, Mesones I, Kembel SW . (2010). A floristic study of the white-sand forests of Peru. Ann Missouri Botanical Garden 97: 283–305.
Fine PVA, Kembel SW . (2011). Phylogenetic community structure and phylogenetic turnover across space and edaphic gradients in western Amazonian tree communities. Ecography 34: 552–565.
Finlay BJ . (2002). Global dispersal of free-living microbial eukaryote species. Science 296: 1061–1063.
Friesen ML, Porter SS, Stark SC, von Wettberg EJ, Sachs JL, Martinez-Romero E . (2011). Microbially mediated plant functional traits. Annu Rev Ecol Evol Systematics 42: 23–46.
Galante TE, Horton TR, Swaney DP . (2011). 95% of basidiospores fall within one meter of the cap- a field and modeling based study. Mycologia 103: 1175–1183.
Gardes M, Bruns T . (1993). ITS primers with enhanced specificity for basidiomycetes—application to the identification of mycorrhizae and rusts. Mol Ecol 2: 113–118.
Gaston KJ . (2000). Global patterns in biodiversity. Nature 405: 220–227.
Gilbert GS, Ferrer A, Carranza J . (2002). Polypore fungal diversity and host density in a moist tropical forest. Biodiversity Conservation 11: 947–957.
Gilbert GS, Sousa WP . (2002). Host specialization among wood-decay polypore fungi in a Caribbean mangrove forest. Biotropica 34: 396–404.
Gilbert GS, Webb CO . (2007). Phylogenetic signal in plant pathogen-host range. PNAS 104: 4979–4983.
Gilbert GS . (2002). Evolutionary ecology of plant diseases in natural ecosystems. Annu Rev Phytopathol 40: 13–43.
Gillett JB . (1962). Pest pressure, an underestimated factor in evolution. Systematics Assoc Publ 4: 37–46.
Green JL, Holmes AJ, Westoby M, Oliver I, Briscoe D, Dangerfield M et al (2004). Spatial scaling of microbial eukaryote diversity. Nature 432: 747–750.
Gullison RE, Frumhoff PC, Canadell JG, Field CB, Nepstad DC, Hayhoe K et al (2007). Tropical forests and climate policy. Science 316: 985–986.
Hansen EM, Goheen EM . (2000). Phellinus weirii and other native root pathogens as determinants of forest structure and process in western North America. Annu Rev Phytopathol 38: 515–539.
Harms KE, Wright SJ, Calderon O, Hernandez A, Herre EA . (2000). Pervasive density-dependent recruitment enhances seedling diversity in a tropical forest. Nature 404: 493–495.
Hawksworth D . (1991). The fungal dimension of biodiversity—magnitude, significance and conservation. Mycol Res 95: 641–655.
Hawksworth DL . (2012). Global species numbers of fungi: are tropical studies and molecular approaches contributing to a more robust estimate? Biodiversity Conservation 21: 2425–2433.
Henkel TW, Terborgh J, Vilgalys RJ . (2002). Ectomycorrhizal fungi and their leguminous hosts in the Pakaraima Mountains of Guyana. Mycol Res 106: 515–531.
Horner-Devine MC, Lage M, Hughes JB, Bohannan BJM . (2004). A taxa-area relationship for bacteria. Nature 432: 750–753.
Hubbell SP . (2001) The Unified Neutral Theory of Biodiversity and Biogeography. Princeton University Press: Princeton, NJ, USA.
Ihrmark K, Bodeker IT, Cruz-Martinez K, Friberg H, Kubartova A, Schenck J et al (2012). New primers to amplify the fungal ITS2 region—evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol Ecol 82: 666–677.
Izzo A, Agbowo J, Bruns TD . (2005). Detection of plot-level changes in ectomycorrhizal communities across years in an old-growth mixed-conifer forest. New Phytologist 166: 619–630.
Janzen DH . (1970). Herbivores and the number of tree species in tropical forests. Am Nat 104: 501–528.
Klironomos JN . (2002). Feedback with soil biota contributes to plant rarity and invasiveness in communities. Nature 417: 67–70.
Knight R, Reeder J . (2010). Denoising pyrosequencing reads by flowgram clustering. Nat Methods 7: 668–669.
Koleff P, Gaston KJ, Lennon JJ . (2003). Measuring beta diversity for presence-absence data. J Anim Ecol 72: 367–382.
Liu X, Ligan M, Etienne RS, Wang Y, Staehlin C, Yu S . (2012). Experimental evidence for a phylogenetic Janzen–Connell effect in a subtropical forest. Ecol Lett 15: 111–118.
Mangan SA, Schnitzer SA, Herre EA, Mack KML, Valencia MC, Sanchez EI et al (2010). Negative plant-soil feedback predicts tree-species relative abundance in a tropical forest. Nature 466: 752–755.
Marengo J . (1998). Climatologia de la zona de Iquito, Peru. In: Kailliola R, Flores Paitan S, Geoecologia y desarrollo Amazonico: estudio integrado en la zona de Iquitos, Peru. University of Turku Press: Turku, pp 35–57.
May R . (1991). A fondness for fungi. Nature 352: 475–476.
McGuire KL, Fierer N, Bateman C, Treseder KK, Turner BL . (2012). Fungal community composition in neotropical rain forests: the influence of tree diversity and precipitation. Microb Ecol 63: 804–812.
Peay KG, Kennedy PG, Bruns TD . (2008). Fungal community ecology: a hybrid beast with a molecular master. BioScience 58: 799–810.
Peay KG, Kennedy PG, Davies SJ, Tan S, Bruns TD . (2010). Potential link between plant and fungal distributions in a dipterocarp rainforest: community and phylogenetic structure of tropical ectomycorrhizal fungi across a plant and soil ecotone. New Phytologist 185: 529–542.
Peay KG, Schubert MG, Nguyen NH, Bruns TD . (2012). Measuring ectomycorrhizal fungal dispersal: macroecological patterns driven by microscopic propagules. Mol Ecol 16: 4122–4136.
Philips OL, Vargas PN, Monteagudo AL, Cruz AP, Zans MC, Sanchez WG et al (2003). Habitat association among Amazonian tree species: a landscape-scale approach. J Ecol 91: 757–775.
Queloz V, Sieber TN, Holdenrieder O, McDonald BA, Grünig CR . (2011). No biogeographical pattern for a root-associated fungal species complex. Global Ecol Biogeography 20: 160–169.
R Core Development Team (2009) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria.
Smith ME, Douhan GW, Rizzo DM . (2007). Intra-specific and intra-sporocarp ITS variation of ectomycorrhizal fungi as assessed by rDNA sequencing of sporocarps and pooled ectomycorrhizal roots from a Quercus woodland. Mycorrhiza 18: 15–22.
Smith ME, Henkel T, Aime MC, Fremier AK, Vilgalys R . (2011). Ectomycorrhizal fungal diversity and community structure on three co-occurring leguminous canopy tree species in a Neotropical rainforests. New Phytologist 192: 699–712.
Smith ME, Henkel TW, Uehling JK, Fremier AK, Clarke HD, Vilgalys R . (2013). The ectomycorrhizal fungal community in a Neotropical forest dominated by the endemic dipterocarp Pakaraimaea dipterocarpacea. PLoS One 8: e55160.
Smith SE, Read DJ . (2008) Mycorrhizal Symbiosis 3rd edn. Elsevier: San Francisco, CA, USA.
Suh SO, Blackwell M, Kurtzman CP, Lachance MA . (2006). Phylogenetics of Saccharomycetales, the ascomycete yeasts. Mycologia 98: 1006–1017.
Tedersoo L, Mohammad B, Toots M, Diedhiou A, Henkel T, Kjoller R et al (2012). Towards global patterns in the diversity and community structure of ectomycorrhizal fungi. Mol Ecol 21: 4160–4170.
Tedersoo L, Nilsson RH, Abarenkov K, Jairus T, Sadam A, Saar I et al (2010). 454 Pyrosequencing and Sanger sequencing of tropical mycorrhizal fungi provide similar results but reveal substantial methodological biases. New Phytologist 188: 291–301.
Terborgh J . (2012). Enemies maintain hyperdiverse tropical forests. Am Nat 179: 303–314.
Toju H, Tanabe AS, Yamamoto S, Sato H . (2012). High-coverage ITS primers for the DNA-based identification of Ascomycetes and Basidiomycetes in environmental samples. PLoS ONE 7: e40863.
Tuomisto H, Ruokolainen K, Yli-Hall M . (2003). Dispersal, environment, and floristic variation of western Amazonian forests. Science 299: 241–244.
van der Heijden MGA, Klironomos JN, Ursic M, Moutoglis P, Streitwolf-Engel R, Boller T et al (1998). Mycorrhizal fungal diversity determines plant biodiversity, ecosystem variability and productivity. Nature 396: 69–72.
Waldrop MP, Zak DR, Blackwood CB, Curtis CD, Tilman D . (2006). Resource availability controls fungal diversity across a plant diversity gradient. Ecol Lett 9: 1127–1135.
Wright SJ . (2002). Plant diversity in tropical forests: a review of mechanisms of species coexistence. Oecologia 130: 1–14.
Acknowledgements
Support for KGP was provided by the National Science Foundation Grant DBI 1045658 and for PVAF and CB by a collaborative NSF Grant DEB-0743800/0743103. We thank Italo Mesones, Julio Sanchez, Julio Grandez Ríos, Tracy Misiewicz, Seth Kauppinen and Fabio Casado for help in field work and comments on previous versions of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Peay, K., Baraloto, C. & Fine, P. Strong coupling of plant and fungal community structure across western Amazonian rainforests. ISME J 7, 1852–1861 (2013). https://doi.org/10.1038/ismej.2013.66
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.66
Keywords
This article is cited by
-
Rhizosphere fungal community assembly varied across functional guilds in a temperate forest
Ecological Processes (2023)
-
Landscape-scale mapping of soil fungal distribution: proposing a new NGS-based approach
Scientific Reports (2023)
-
Effects of close-to-nature management of plantation on the structure and ecological functions of soil microorganisms with different habitat specialization
Plant and Soil (2023)
-
Plant and fungal species interactions differ between aboveground and belowground habitats in mountain forests of eastern China
Science China Life Sciences (2023)
-
High variation of fungal communities and associated potential plant pathogens induced by long-term addition of N fertilizers rather than P, K fertilizers: A case study in a Mollisol field
Soil Ecology Letters (2022)
