
Abstract
Snow environments can occupy over a third of land surface area, but little is known about the dynamics of snowpack bacteria. The effect of snow melt on bacterial community structure and diversity of surface environments of a Svalbard glacier was examined using analyses of 16S rRNA genes via T-RFLP, qPCR and 454 pyrosequencing. Distinct community structures were found in different habitat types, with changes over 1 week apparent, in particular for the dominant bacterial class present, Betaproteobacteria. The differences observed were consistent with influences from depositional mode (snowfall vs aeolian dusts), contrasting snow with dust-rich snow layers and near-surface ice. Contrary to that, slush as the decompositional product of snow harboured distinct lineages of bacteria, further implying post-depositional changes in community structure. Taxa affiliated to the betaproteobacterial genus Polaromonas were particularly dynamic, and evidence for the presence of betaproteobacterial ammonia-oxidizing bacteria was uncovered, inviting the prospect that the dynamic bacterial communities associated with snowpacks may be active in supraglacial nitrogen cycling and capable of rapid responses to changes induced by snowmelt. Furthermore the potential of supraglacial snowpack ecosystems to respond to transient yet spatially extensive melting episodes such as that observed across most of Greenland’s ice sheet in 2012 merits further investigation.
Similar content being viewed by others
Introduction
Most of our biosphere is permanently cold (Morita, 1975; Russell, 1990). Among these cold environments, snow permanently or seasonally covers up to 35% of the Earth’s terrestrial surface area and drives powerful feedbacks within the climate system (Miteva, 2008).
Microbes have important roles in snow ecosystems, as detailed in reviews of snow ecology (Jones, 1999; Hoham and Duval, 2001). Snow algal blooms are well known as prominent loci of primary production in nival ecosystems (Hoham and Duval, 2001; Takeuchi, 2001). However, although metabolically active bacteria are present in snowpacks the world over, even in remote locations such as the South Pole (Carpenter et al., 2000), these are less studied. In Alpine snowpacks, bacterial abundances range between 103–105 cells ml−1 (Alfreider et al., 1996; Sattler et al., 2001). Bacteria in snowpacks may be inoculated by precipitation and dry deposition (Xiang et al., 2009b), however the relative importance of depositional and post-depositional processes in structuring bacterial communities in different environments is less clear. Seasonal shifts in snowpack bacterial communities have been reported in mountainous regions of Japan (Segawa et al., 2005), as increased snow temperature and meltwater content during seasonal melt promote rapid microbial growth, but also induce washout of cells by elution in meltwater, leading to seasonal shifts in snowpack bacterial communities.
In Svalbard, snow bacterial abundances of about 2 × 104 cells ml−1 and the cultivation of Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria, Firmicutes and Actinobacteria has been described (Amato et al., 2007). Previous clone library studies of High Arctic snow have identified the predominance of Betaproteobacteria in snowpacks containing 19 different classes of bacteria (Larose et al., 2010) and six different phyla (Harding et al., 2011). The sequences retrieved were typically related to other sequences from cold environments, although the potential for spatial and temporal variation in snowpack microbial diversity at fine scales of resolution were less well explored.
Nevertheless, interactions between Arctic snowpack communities and environmental changes are currently attracting increasing attention (Larose et al., 2013). As well as impacts on seasonal melting from climate shifts, other impacts include the potential transformation of mercury (Møller et al., 2011) and other pollutants. Atmospheric deposition of nitrogen (Gjessing, 1977; Simoes and Zagorodnov, 2001; Erisman et al., 2008) is important in otherwise pristine Arctic environments. Glacial ecosystems can be influenced by episodic nitrogen deposition events, which may become more frequent in a changing Arctic (Hodson et al., 2010; Roberts et al., 2010; Kühnel et al., 2011). Furthermore, it is thought that microbially mediated nitrification may occur within the snowpack itself (Amoroso et al., 2010) and ammonia-oxidizing Betaproteobacteria are active nitrifiers in glacial ice microcosms (Miteva et al., 2007), and nifH genes indicate the potential for nitrogen fixation in supraglacial snow (Boyd et al., 2011). Finally, significant increases in anthropogenic nitrogen inputs to Svalbard and other glacierized regions (Döscher et al., 1996; Fischer et al., 1998; Kekonen et al., 2005) modulated via supraglacial snowpacks affect the nitrogen cycle (Telling et al., 2011) of supraglacial hotspots of biodiversity and activity such as cryoconite holes (Edwards et al., 2011).
In the light of our limited understanding of Arctic snowpack bacterial communities that may prove significant in terms of spatial extent, biogeochemical cycling and sensitivity to a changing climate, greater insight into the distribution and dynamics of snowpack communities is needed. A particular lacuna in our knowledge is how seasonal melting, the extent, rate and duration of which is affected by a changing climate, affects these microbial communities.
In this study, samples collected from a High Arctic glacier’s snowpack during seasonal melting were subjected to T-RFLP, pyrosequencing and qPCR to examine the bacterial community and betaproteobacterial ammonia-oxidizers. The primary objective was to evaluate whether habitats associated with the supraglacial snowpack of a High Arctic glacier harboured bacterial communities that could rapidly respond to changes induced by advancing melting.
Materials and methods
Study site and sampling
To test for changes in the snowpack bacterial community at advancing stages of melting, samples were taken on the 6th and 11th of July 2010 (hereafter, Day 1 and Day 2) on Larsbreen, which is a High Arctic polythermal valley glacier near Longyearbyen (Svalbard, 78° 13′N, 15° 38′E; Figure 1). The glacier is 2800 m long and 700 m wide, with an area of 2.96 km2 (Etzelmüller et al., 2000).

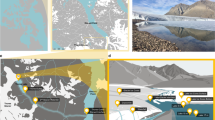
Study location and design. (a) Overview of the glacier’s location. Larsbreen is a High Arctic valley glacier near Longyearbyen, Svalbard (inset). (b) At each sampling station, three snowpits were at the corners of a triangle 10 m apart; snowpack profiles and the range of surface snow, snow and slush habitat thicknesses illustrated. (c) shows the locality of sampling stations 1, 2 and 3, where, triplicate snowpits were excavated as described.
Three sample stations 1 km apart (1, 2 and 3) along an altitudinal transect on the glacier were chosen (Figures 1 and 2) to sample across a gradient of melting season duration and intensity. On each side, three pits (A, B and C), 10 m away from each other, were dug to measure snow- and slush depth.

qPCR estimation of mean β-AOB 16S rRNA gene abundance in different habitats; error bars represent ±1 s.e.m.
Samples from the snowpack, slush and the underlying near-surface ice (Figure 1) were collected. Due to high amounts of aeolian depositions observed on the second day of sampling, surface snow (1 cm depth) was additionally sampled. Further details of the sampling protocol, chemical and microbial activity analysis are provided as Supplementary Methods.
Within 24 h of sampling, 1.5–2 l sample volume was filtered through sterile 0.2-μm pore-size polycarbonate filters (Millipore Ltd., Watford, UK) at the laboratory in Longyearbyen. Filters were transported frozen on dry ice to Austria and stored at −20 °C until DNA was extracted.
In addition to sampling, snow depths were taken on five spots in the close vicinity of each pit, together with the measurement of slush depths. Conductivity and pH were measured on day 1.
DNA extraction and PCR amplification
DNA was extracted from membrane filters using the MoBio, Inc., PowerSoil DNA extraction kit as directed by the manufacturer (MoBio, Inc., Solana, CA, USA), with the exceptions that following bead-beating, three freeze–thaw cycles were conducted (30 min at −80 °C followed by 5 min at 65 °C each time) and DNA was eluted in 10 μl of buffer C6. All pre-PCR manipulations were conducted in sterile conditions on bleach-disinfected work surfaces in laminar flow hoods in rooms not used for handling amplified sequences, using filter tips and certified DNA- and DNase-free plasticware. PCR mixtures used for amplifications contained 2.5 μl of 10 × Taq buffer with KCl, 200 μM each of deoxynucleoside triphosphate, 0.24 μM of each primer, 1.5 mM MgCl2, 0.2% bovine serum albumin, 0.625U Taq DNA Polymerase (Fermentas, York, UK), 2 μl of extracted DNA and nuclease-free water to a volume of 25 μl. Details of all PCR primers used in the study are specified as Supplementary Table 1.
Terminal restriction fragment length polymorphism (T-RFLP)
T-RFLP was conducted generally as described in Edwards et al. (2011), with the reaction components described above for primers Cy5-27F and 1389R for the bacterial 16S ribosomal RNA gene.
To target betaproteobacterial ammonia-oxidizing bacteria (hereafter β-AOB), a T-RFLP assay based upon a 465-bp β-AOB 16S rRNA gene PCR product was amplified using an equimolar mixture of the two forward primers CTO189f A+B and CTO189f C, 5′-tagged with fluorophore Cy5 (5-carboxyfluorescein), and a reverse primer CTO654r based upon frequently used β-AOB-specific denaturing gradient gel electrophoresis (Kowalchuk et al., 1997) modified so as to exclude the GC-clamp. In the instance of β-AOB T-RFLP, PCRs were conducted for 40 cycles on 27F/1389R templates to assure ample product for T-RFLP.
For both T-RFLP analyses, PCR products were cleaned, digested with 5U HaeIII (Promega, Inc., Madison, WI, USA) for 4.5 h, spin column-purified and the fragments were analyzed using a Beckmann-Coulter CEQ8000 (Beckmann-Coulter, High Wycombe, UK) exactly as described by Edwards et al. (2011). Analysis of terminal restriction fragment relative abundance profiles was conducted with PERMANOVA (Anderson, 2001) using PRIMER6 and PERMANOVA+ (version 1.0.2 and version 6.1.12; Primer-E, Ivybridge, UK) and CCA (ter Braak, 1986) using MVSP 3.1 (Kovach Computing Services, Sir Fôn, UK), respectively, and was carried out as previously described (Edwards et al., 2011).
Real-time quantitative PCR (qPCR) of β-AOB partial 16S rRNA genes
To explore variation in the abundance of β-AOB genes in the sample types collected, 2 μl of each DNA extract was subjected to real-time PCR in 20-μl reactions using a SYBR Green I ABSolute qPCR chemistry (ABGene, Epsom, UK) in 40 cycles as described above using a Bio-Rad iQ Cycler (Bio-Rad, Ltd., Hemel Hempsted, UK) and the β-AOB 16S rRNA gene primers described in Supplementary Table 1. A plasmid standard bearing the 465-bp β-AOB fragment derived from PCR of DNA from cryoconite sampled in the European Alps (AEdwards and B Sattler, unpublished data) cloned as described below was extracted using a QIAQuick plasmid spin minprep kit (Qiagen, Ltd, Crawley, UK) and prepared as a 1:10 dilution series to represent 10–100 000 copies of the β-AOB fragment. A PCR efficiency of 93.7%, and gene copy number per extract was estimated by the iQ cycler’s software. All DNA extracts were used in this study were tested by qPCR. As our objective was the estimation of β-AOB 16S rRNA copy number, and the volume of each DNA extract available was very limited in availability, normalisation to total 16S rRNA gene abundances was not possible. Details of cloning are provided in Supplementary Methods.
454 amplicon pyrosequencing
To complement profiles of community structure by deeply sequencing the bacterial 16S rRNA gene, 454 amplicon pyrosequencing was performed on samples from each habitat type. As barcoded amplicons could not be generated directly from DNA extracts (data not shown), a hemi-nested PCR approach using 15 cycles of PCR with primers 27F and 1389R, as described above, was required prior to amplification using barcoded primers. In the second round of amplification, the primers consisted of 27F and 357R (5′-tagged with the Roche A and MID barcode tags). One microlitre of PCR product was used in 25-μl reactions containing 10 × reaction buffer with1.8 mM MgCl2, 0.2 mM dNTPs, 0.2 μM of each primer, 1.25 U FastStart High Fidelity Enzyme Blend (Roche Biosystems, Burgess Hill, UK), and subjected to 30 cycles of 30 s at 95 °C, 30 s at 55 °C and 2 min at 72 °C and terminated by a single final elongation of 7 min at 72 °C.
Amplicons were cleaned using Agencourt AMPure XP beads (Beckman-Coulter Genomics) prior to pyrosequencing using Titanium chemistry and protocols on the Aberystwyth University Roche GS-FLX 454 sequencer (Roche Diagnostics Ltd). Sequences were de-multiplexed, quality-filtered and analyzed using QIIME (Caporaso et al., 2010). Operational taxomomic units (OTUs) were assigned using UCLUST at a threshold of 97% pairwise identity, and representative sequences from each OTU were selected for taxonomy assignment. These sequences were aligned using PyNAST and classified using the Ribosomal Database Project classifier against the RDP 16S rDNA core set using a 0.80 confidence threshold. A matrix of each OTU’s relative abundance in each sample was imported into PRIMER6 and PERMANOVA+ for PERMANOVA and CAP (Anderson and Willis, 2003) and calculation of richness and Shannon–Wiener diversity index. Univariate statistics were conducted in Minitab 12.23 (Minitab, Ltd., Coventry, UK).
Results
Samples of snow (n=15), surface layer snow (n=16), slush (n=15) and ice (n=14) were collected for molecular and chemical analysis on both Day 1 and Day 2; summary details of snowpack properties and chemistry are provided in Table 1 and Supplementary Figure 1. As can be determined from Table 1, the decrease in snowpack depth and increase in the slush fraction illustrate the advancing melting of the snowpack in the interim between Day 1 and Day 2.
Bacterial community structure of the Larsbreen snowpack
DNA was successfully extracted from all 60 samples of snow, slush and ice, permitting T-RFLP analyses of bacterial and β-AOB 16S rRNA genes. Profiles meeting quality filtering criteria were generated from all samples, with the exception of four samples subjected to bacterial 16S rRNA gene T-RFLP (Slush 2B and 2C from Day 1, Ice 1B III+IV as well as Ice 2B III+IV from Day 2).
To test the hypotheses that (i) different habitat types (seasonal snow-pack, slush and near-surface ice) harbour distinct communities and (ii) these communities are subject to change as the melting season progresses, Bray–Curtis distances of fourth-root transforms of T-RFLP profiles (thus indicative of differences in community structure) were analysed by PERMANOVA to test for significant differences. A three-factor design to test for differences between sampling station, time and habitat type was conducted for both bacterial and β-AOB-specific profiles.
For both bacterial and β-AOB T-RFLP data sets, no significant difference was observed between sampling stations (Pseudo-F=1.08, p(perm)=0.366 and Pseudo-F=0.911, p(perm)=0.522, respectively). However, significant differences were returned for both communities due to time, consistent with a change in the structure of both bacterial and β-AOB communities (Day 1 vs Day 2; Pseudo-F=5.08, p(perm)=0.0016 and Pseudo-F=2.97, p(perm)= 0.0149, respectively). Equally, for both communities, highly significant differences according to habitat type were apparent (F=3.56, p(perm)= 0.0001 and Pseudo-F=4.13, p(perm)=0.0001, respectively) for bacteria, in general, and β-AOB. Pairwise PERMANOVA tests between habitats revealed significant differences between habitats for both bacterial and β-AOB 16S rRNA gene T-RFLPs, with the exception of snow surface and ice samples for bacteria and snow and slush for β-AOB at the P=0.05 level. Details are provided in Table 2. No interactions between factors (time, habitat type or sampling station) were apparent (P>0.1 in all instances). In summary, our T-RFLP results are consistent with the presence of temporally dynamic bacterial and betaproteobacterial ammonia-oxidizer communities partitioned between distinct habitats throughout the melting snowpack of Larsbreen.
Although the primers used to amplify β-AOB 16S rRNA genes have been frequently used to specifically profile β-AOB communities using DGGE (Kowalchuk et al., 1997), yielding high levels of β-AOB diversity (Mahmood et al., 2006), and T-RFLP results were broadly congruent with DGGE profiles (for both bacteria, in general, and β-AOB 16S rRNA genes; (Supplementary Figures 2–5), we sought to verify the specificity of the β-AOB T-RFLP method given that we believe this is the primers’ first application for T-RFLP. Therefore, qPCR products from all samples were pooled and cloned into pGEM T-Easy, and clones yielding PCR products consistent with the presence of an insert were sequenced. Of the clones successfully aligned to the RDP taxonomic hierarchy (28/32 clones; Genbank HF934045—HF934072), these all lay within three clades of Betaproteobacteria in neighbour-joining trees, and the best matches using BLASTN were for uncultivated β-AOB, typically from cold environments (Supplementary Figure 6 and Supplementary Table 2). Thus, variation in T-RFLP profiles and qPCR results between habitats is likely to reflect variation in β-AOB community structure and abundance. This is amply illustrated by qPCR results, with mean β-AOB 16S rRNA gene copies per microlitre of DNA extract varying between 216 copies per microlitre of extract in the snow surface and 72 000 copies per microlitre of extract in near-surface ice (Figure 3).

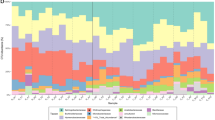
Barcoded amplicon 454 pyrosequencing of bacterial 16S rRNA genes. (a) Canonical Analysis of Principal Components of UCLUST 97%-clustered OTUs modelled by habitat type (total misclassification error=20%; slush: hollow triangles, snow: filled triangles, surface snow: squares, ice: diamonds.) (b) distribution of OTUs aligned and assigned to known bacterial phyla as the cumulative relative abundance of OTUs associated with each phylum. (c) Percentage of OTUs affiliated to known bacterial phyla.
Bacterial diversity of the Larsbreen snowpack
Twenty DNA samples representing at least three randomly selected samples each from snow and slush on both Day 1 and Day 2, as well as four surface snow and three ice core samples were subjected to V1–V3 16S rRNA gene amplicon pyrosequencing. Attempts to amplify 16S rRNA gene fragments directly from community DNA using 27F–357R were unsuccessful, potentially due to challenges from amplifying from low-biomass samples with tagged primers. A hemi-nested approach using 27F–1389R as template for the barcoded primer PCR reaction robustly amplified specific products with no contamination apparent in negative controls. This approach has the advantage of minimizing bias associated with direct amplification with barcoded primers (Berry et al., 2011). From this analysis, 254 262 reads successfully passed the QIIME pipeline at an abundance of ⩾5 reads within the data set. The full 454 amplicon dataset is available at EBI-SRA (ERP002433).
These reads constituted 818 operational taxonomic units at the 97% sequence identity level. No statistical difference was apparent according to sample type in terms of richness (mean of 333 OTUs per sample±84.1, 1 s.d.; one-way ANOVA; F=2.17, P=0.132). However, significant differences were apparent in terms of Shannon–Wiener diversity indices (one-way ANOVA, F=5.30, P=0.01), accounted for by significantly higher diversity in ice samples compared with snow and slush according to post-hoc Tukey testing; no difference was apparent between surface snow samples and other habitat types. Likewise, significant differences were apparent between habitat types using a single-factor design PERMANOVA (pseudo-F=3.58, p(perm)=0.0001) of Bray–Curtis distances of fourth-root transforms of OTU relative abundances.
Pairwise PERMANOVA comparisons reveal differences between most habitat types with the exception of comparisons of snow surface and snow habitats, which achieves borderline significance (P=0.07), and between snow surface and ice (P=0.102); these results are summarized in Table 3. Figure 3a illustrates the ordination by CAP of the 454 profiles according to the habitat-type model. This correctly classifies 16/20 samples to the correct habitat type, with the greatest source of error associated with snow surface samples, with one sample each distributed to snow, snow surface and ice habitats. Overall, the robustness of the model specifying habitat-specific communities is supported.
Of the 454 OTUs assigned to phyla by alignment to the RDP core set at a confidence of ⩾0.8, at least seven phyla are present (Figure 3). Proteobacteria overwhelmingly dominate the communities of all habitat types sequenced. Within the phylum Proteobacteria, the class Betaproteobacteria are highly abundant in snow and slush in terms of relative abundance of reads, but a broadly even distribution of proteobacterial richness is evident across samples. Alignment of 454 OTUs to the Greengenes taxonomy at a confidence level of 0.5 resulted in the alignment of more OTUs to taxonomy, but these OTUs were predominantly mapped to plastid sequences, and in particular, chloroplast 16S rRNA genes affiliated with Chlamydomonadaceae, which include the snow algal taxa. Exclusion of plastid-derived OTUs yielded similar results using the GreenGenes taxonomy to stringent alignment to the RDP core set, with Betaproteobacteria proving abundant, particularly within snow and slush. (Greengenes-assigned Betaproteobacteria OTUs: slush 56%; snow, 36%; snow surface, 24%; ice 24% mean relative abundance, one-way ANOVA, F=6.84, P=0.004). These results are shown as Supplementary Figure 7. Consequently, we limited further analysis to RDP-aligned taxa.
Further inspection of RDP-based OTU assignments reveal that 28 OTUs affiliated to a single genus, Polaromonas, are significant contributors to this abundance. This is most apparent for a single Polaromonas OTU, OTU#1649, which accounts for 1.9–45.8% of all reads per sample in the data set (habitat means: 11.8% for snow, 27.8% for slush, 3.9% for surface snow and 8.4% for ice). OTU#1649 is present at significantly higher abundance in slush habitats compared with all other habitat types (one-way ANOVA with post-hoc Tukey F=7.10, P=0.003) and remains stable within the slush habitat over time with no interactions between habitat and time (general linear model ANOVA: slush vs snow, Day 1 vs Day 2; habitat × time. Habitat: F=10.29, P=0.009; Time: F=1.21, P=0.297; habitat × time F=1.26, P=0.288; Supplementary Figure 8). A similar trend is apparent for the relative abundance of all Polaromonas affiliated OTUs (general linear model ANOVA: slush vs snow, Day 1 vs Day 2; habitat × time. Habitat: F=10.91, P=0.008; time: F=3.37, P=0.096; habitat × time F=0.76, P=0.404) However, excluding the influence of OTU#1649 on Polaromonas-affiliated OTU relative abundances diminishes this trend (general linear model ANOVA: slush vs snow, Day 1 vs Day 2; habitat × time. Habitat: F=4.76, P=0.054; Time: F=8.88, P=0.014; Habitat × time F=0.02, P=0.890). This would lend support to the hypothesis that this particular OTU is capable of responding to the emergence of a seasonal slush layer, and persist at considerable abundance within this particular habitat.
To assess the responses of Polaromonas-affiliated OTUs as an assemblage of taxa, PERMANOVA analysis of Bray–Curtis distances of fourth-root transforms of OTU relative abundances was conducted. This returned highly significant differences between Polaromonas community structure according to habitat (pseudo-F=4.86, P=0.0013), and a significant difference due to time (pseudo-F=3.68, P=0.0157), without interaction between these main effects (pseudo-F=1.01, P=0.396). The highly significant difference between habitat types is predominantly due to differences between slush and other habitat types (pairwise PERMANOVA P-values in the range 0.001–0.03 for all comparisons with slush), with the only other difference at the P<0.05 level being between the snow and the snow surface (P=0.02).
Due to the apparent abundance of Polaromonas-affiliated OTUs, to verify whether the peculiar distribution of this genus was predominantly responsible for the patterns of taxon distribution observed, PERMANOVA analyses of all OTUs and all beta-proteobacterial affiliated OTUs were repeated, with the Polaromonas-affiliated OTUs excluded from the analysis. These analyses confirmed the robustness of overall patterns in OTU and beta-proteobacterial OTUs from Polaromonas influences. Using the above model, PERMANOVA returned similarly significant differences for the full data set and both the full data set and beta-proteobacterial data sets exclusive of Polaromonas-affiliated OTUs; these are summarized in Table 3. In conclusion, although Polaromonas-affiliated OTUs demonstrate a distinctive distribution within the habitats sampled, other taxa are involved in structuring the bacterial communities of the glacier’s melting snowpack.
Indeed, as highlighted in Table 3, such variation by habitat is apparent for a range of higher-order taxa. Of the seven phyla (including five classes of Proteobacteria), all phyla and three of the proteobacterial classes (with the exception of Deltaproteobacteria and Epsilonproteobacteria with two OTUs each) were present to a sufficient degree of OTU richness to permit multivariate analyses of their distribution. These results are reported in Table 3. It is noticeable that with the exception of the Firmicutes and TM7 phyla, all others groups of taxa demonstrate significant differences between habitats at the P<0.05 level. However, only the Betaproteobacteria demonstrate significant temporal variation (pseudo-F=4.33, p(perm)=0.0077) between Day 1 and Day 2. Therefore, while the influence of habitat type are apparent on the snowpack bacterial communities, as evidenced by significant differences in the partitioning of higher-order taxon diversity, the Betaproteobacteria prove to be the most dynamic members of the snowpack bacterial community of a High Arctic glacier.
Habitat and microbial activity interactions with community structure
In the light of the shifts in community structure revealed by molecular analyses, attempts to relate the effect of snowpack chemistry and bacterial carbon production (as determined by [3H]leucine incorporation, to be fully reported elsewhere by J Zarsky, K Hell, R Psenner and B Sattler) on community dynamics were made. Canonical correspondence analysis of these parameters’ influence on the relative abundance of OTUs assigned to the Ribosomal Database Project (RDP) hierarchy discriminated slush habitats from snow and snow surface layers clearly, (Figure 4) with a stepwise-optimized model that explained 63.6% of total variance within the first three axes. The effect of calcium, chloride, nitrate and sulphate concentrations on slush communities is apparent. Examination of intraset correlations (Supplementary Table 4) reveals the influence of total nitrogen and bacterial carbon production as the strongest influences on the first environmental axis, followed by ammonium concentrations. These patterns are broadly congruent with CCA conducted similarly using both T-RFLP data sets (Supplementary Figure 9), where the influence of ammonium, total nitrogen, bacterial carbon production and dissolved organic carbon concentrations are apparent in intraset correlations (Supplementary Table 3). Changes in the properties of the slush habitat were investigated further by ANOVA to provide insight into the dynamic habitat occupied by slush bacteria. No significant spatial effects on environmental parameters concentrations were identified, but significant decreases were observed in the abundance of several parameters. Concentrations of chloride (F=5.73, P=0.032), nitrate (F=6.07, P=0.028), sulphate (F=6.02, P=0.029), magnesium (F=6.72, P=0.022) and calcium (F=7.90, P=0.015) decreased significantly in slush between Day 1 and Day 2, with a borderline significant decrease in sodium concentration (F=4.81, P=0.047) also apparent. No significant changes in ammonium, potassium, dissolved organic carbon, total nitrogen or bacterial carbon production were detected.

Canonical correspondence analysis model of environmental variable influence on the relative abundance of RDP-aligned OTUs. The variables displayed were retained following stepwise optimization of multicollinearity. Percentage of total variance explained per environmental axis is indicated for both axes. Supplementary Table 4 provides further details.
Discussion
The presence of a diverse microbial community associated with snow environments found all over the world, including the High Arctic, is increasingly recognized (Carpenter et al., 2000; Amato et al., 2007; Xiang et al., 2009a; Larose et al., 2010; Chuvochina et al., 2011; Harding et al., 2011). Although the potential for depositional and post-depositional processes to influence the structure of these microbial communities is proposed (Xiang et al., 2009b), our understanding of the scale of spatial and temporal variation of snow community composition is limited, in spite of the extensive global areal extent of snow cover. This study therefore offers the first quantitative evidence from 16S rRNA gene-based community structure profiling and amplicon pyrosequencing that different snowpack-associated habitats from a High Arctic glacier harbour distinct and diverse bacterial communities, and that these can be temporally dynamic within the melting season.
Little evidence was found of spatial variation in the bacterial communities between sampling stations covering gradients of elevation (200 m) and 2 km horizontal distance. However, at each sampling station, clear effects of habitat type are apparent in influencing the bacterial communities of surface snow, snow, slush and near-surface ice. Most phyla present (Acidobacteria, Actinobacteria, Bacteroidetes, Cyanobacteria and Proteobacteria) show significantly different habitat distributions (whereas, Firmicutes and the TM7 candidate phylum do not) as revealed by PERMANOVA (Table 3). Differences between snow habitats, slush and ice account for variations in the distribution of OTUs affiliated to these phyla.
Within these comparisons, it is interesting to note the resemblance between surface snow samples and ice cores consistently revealed by both T-RFLP analyses and 454 pyrosequencing. In one respect, these results are unexpected, considering the contrasting nature of these habitats, respectively being the most recently formed and the oldest habitats, with surface snow evidently at the atmosphere–snowpack interface, and the glacier ice at the interface with the slush layer.
However, these differences may be accounted for by the mode of cell deposition. Surface snow samples consisted of the top centimetre of the snowpack sampled on Day 2, serendipitously exploiting a dust deposition event in the interim as a means of evaluating the dry deposition of cells. It is therefore likely that the ‘surface snow’ bacterial community is strongly influenced by organisms introduced by aeolian (or ‘dry’) deposition. In contrast, snow (and by extension, its decomposition product, slush) constitutes predominantly ‘wet’, or precipitation-related deposition (Xiang et al., 2009b); all analyses differentiate snow and slush with surface snow in terms of community composition. Glacial ice is likely to integrate microbes introduced by both deposition mechanisms, and thus may conserve dry-deposited microbes. Support for this contention is provided by both the association of biomass with dust-rich layers in ice cores (for example, Xiang et al., 2009a). Flow cytometric analyses of cell and particle budgets of High Arctic glaciers (Irvine-Fynn et al., 2012) demonstrate net accumulation of cells and particulates in surface ice. From the amplicon pyrosequencing data presented here, the significantly increased Shannon–Wiener diversity of ice relative to snow and slush, and the lack of a significant difference between ice and surface snow in terms of Shannon–Wiener diversity indices would be consistent with this interpretation.
Both bacterial 16S rRNA gene T-RFLP and 454 amplicon pyrosequencing reveal differences in bacterial community structure and composition between snow and slush layers. As slush constitutes the decomposition product of a melting snowpack, these observations are consistent with a population dynamic occurring in the snowpack community during its melting. This is further supported by the significant effect of time on T-RFLP profiles and 454 amplicon compositions in PERMANOVA analyses. In summary, in the five days between Day 1 and Day 2 sampling points, a considerable shift occurred in the structure of bacterial communities within Larsbreen’s snowpack. Although seasonal influences on the composition of 16S rRNA gene clone libraries from snow have been reported previously, including on High Arctic glaciers (Larose et al., 2010), the results presented herein illustrate a considerable capacity for even rapid change in the bacterial communities associated with snowpacks, thus underlining a role for snow as a dynamic habitat for active microbial communities.
Nevertheless, it is apparent that rapid changes in community structure are restricted mainly within the dominant phylum, Proteobacteria, and particularly the Betaproteobacteria. OTUs affiliated with Betaproteobacteria appear uniquely responsive to the effect of time (Table 3, p(perm) =0.0077), and that this trait appears coherent at the class level, by inference of its retention even when the influence of Polaromonas-affiliated OTUs is accounted for (Table 3, p(perm)=0.0073). The dominance and dynamic nature of betaproteobacterial taxa in the snowpack therefore merits further consideration, especially since its prevalence is consistent with other (clone library) based studies of High Arctic snowpacks (Larose et al., 2010; Harding et al., 2011). In this respect, snowpacks seem comparable to other freshwater systems (Newton et al., 2011).
Although the study of coherence between microbial taxonomy and functionality in the cryosphere is in its infancy, in aquatic and terrestrial systems, Betaproteobacteria are characterized as r-strategists, capable of rapidly responding to environmental fluctuations (Fierer et al., 2007) and the utilization of diverse nutritional sources. In this regard, they resemble ‘opportuni-troph’ organisms (Polz et al., 2006), exceedingly capable of exploiting spatially and temporally variable resources. Therefore, the finding of significant shifts in Betaproteobacteria diversity over a short period of time is likely to be consistent with exploitation of favourable conditions within a melting snowpack. The ionic environment of a melting snowpack is particularly dynamic (Johannessen and Henriksen, 1978). Impurities are differentially eluted from the snowpack in a nonlinear manner (Brimblecombe et al., 1985), as ions are rejected and relocated as snow metamorphoses (Cragin et al., 1996). Such changes are broadly reflected by the chemistry data (Supplementary Figure 1). Variations in the ionic composition of slush, in particular, calcium, chloride, nitrate and sulphate due to elution clearly ordinate slush from snow habitats in the CCA model of environmental influences on OTU abundances (Figure 4). This lends support to the argument that Betaproteobacteria that exploit slush as a habitat must prove exceptionally versatile and tolerant of a dynamic physicochemical environment.
This tendency is illustrated by the distribution of the bacterial genus that appears dominant within the snowpack-associated communities, Polaromonas. OTUs affiliated to this genus constitute a considerable proportion of all 454 reads throughout the data set, with one Polaromonas OTU alone, OTU#1648, accounting for up to 45% of reads in one sample in the most extreme instance. This OTU is present at a significantly higher relative abundance within the slush layer, and persists within the slush layer between Day 1 and Day 2, in contrast to the summed relative abundance of all remaining Polaromonas-affiliated OTUs. It is therefore likely that this taxon is well adapted to life in the seasonal slush layer of the glacier, and thus proliferates rapidly within it, while other members of Polaromonas occupy a broader distribution within the snowpack. The appearance of a high ratio of local to global diversity in Polaromonas, as recently reported (Darcy et al., 2011), would provide opportunities for co-dispersed Polaromonas species to exploit distinct niches in space and time, utilizing their considerable metabolic versatility (Mattes et al., 2008; Yagi et al., 2009) to do so.
Because of the abundance of Betaproteobacteria and isotopic evidence of snowpack nitrification (Amoroso et al., 2010), we sought to evaluate the distribution and dynamics of β-AOB. DGGE analysis (Supplementary Figures 4 and 5), as well as T-RFLP (Table 2) confirmed by qPCR (Figure 2), and a small clone library (Supplementary Figure 6 and Supplementary Table 2) served to demonstrate the presence of β-AOB in all habitats, with different community structures apparent between the snowpack and the ice surface. Coupled with the greater abundance of β-AOB in the ice, these data suggest differing supraglacial distributions of β-AOB. Activity of β-AOB in microcosms representing glacial ice cores suggests that populations accumulated in near-surface ice may be active (Miteva et al., 2007). Amplification of bacterial or archaeal AmoA genes was unsuccessful (data not shown), thus limiting the potential for further insights. PCR detection of AmoA was recently reported in cryoconite habitats on a glacier <20 km from Larsbreen (Cameron et al., 2012). Many other cryoconite holes yielded multimeric PCR products (Cameron et al., 2012), while diverse forms of β-AOB AmoA were reported from subglacial sediments (Boyd et al., 2011). These highlight challenges in detecting functional genes in (low-biomass) glacial environments, possibly accounting for incongruence. Whether the snowpack is an active locus of nitrification remains to be confirmed, but it is certainly possible that β-AOB accumulated in snowpack-associated habitats, if exported (Irvine-Fynn et al., 2012), serves to inoculate other ice surface (Cameron et al., 2012), subglacial (Boyd et al., 2011) or forefield (Brankatschk et al., 2011) habitats, dependent on the route of meltwater (Hodson et al., 2008).
The decoupling of nutrient dynamics from the elution of other snowpack solutes during melt may infer the importance of the snowpack biota in nitrogen budgets (Hodson, 2006), but amendment experiments in alpine environments (Williams et al., 1996) fail to support this contention. The particular influence of total nitrogen and ammonium concentrations collinear with bacterial carbon production rates is apparent in all CCA models of snowpack bacterial communities reported here. This lends further support to the role of snowpack communities in sequestering nitrogen resources, as argued by workers conducting annual budgets of glacierized catchment biogeochemistry (Hodson et al., 2005; Hodson, 2006).
Although this study was performed within a single glacierized catchment on Svalbard, considering the commonality in the groups of taxa observed with other snowpack diversity studies (Amato et al., 2007; Larose et al., 2010; Harding et al., 2011), and that specific groups of interest (that is, Polaromonas; (Darcy et al., 2011)) are thought to be very efficiently dispersed in the cryosphere, it is tempting to generalize that snowpack communities are able to respond rapidly to snowmelt, even on timescales as brief as several days to a week, in particular, as anthropogenic nitrogen deposition also affects other glacial systems (Fischer et al., 1998). As the recent spatially extensive yet transient melting of the Greenland ice sheet snowpack observed on July 2012 (Nghiem et al., 2012) led to extensive saturation of firn on the Greenland ice sheet interior (A Hubbard, personal communication), the potential for melt-induced pulses to initiate snowpack microbial processes during such episodes merits further investigation. Furthermore, as supraglacial runoff incorporates bacteria (Irvine-Fynn et al., 2012) that may influence ice-marginal habitats (Mindl et al., 2007), the impacts of snowpack bacterial community shifts may extend beyond the glacier itself. Betaproteobacteria and, in particular, Polaromonas strains were recently shown to significantly accelerate mineral weathering in an Alpine glacier forefield (Frey et al., 2010), so the prominence of these organisms in slush may influence paraglacial dynamics downstream.
In conclusion, the presence of a diverse and dynamic bacterial community is revealed within the melting snowpack of a High Arctic glacier by multiple molecular methods. Clear differences in community structure according to habitat type are evident, implying the role for both depositional and post-depositional influences in structuring the bacterial community of snowpacks. The dominance and dynamic nature of Betaproteobacteria, even at the brief timescales examined would indicate their potential to proliferate in transient niches, and hence, exploit the dynamic environment of a melting snowpack with consequent impacts upon glacierized catchment biogeochemistry.
Accession codes
References
Alfreider A, Pernthaler J, Amann R, Sattler B, Glockner FO, Wille A et al (1996). Community analysis of the bacterial assemblages in the winter cover and pelagic layers of a high mountain lake by in situ hybridization. Appl Environ Microbiol 62: 2138–2144.
Amato P, Hennebelle R, Magand O, Sancelme M, Delort AM, Barbante C et al (2007). Bacterial characterization of the snow cover at Spitzberg, Svalbard. FEMS Microbiol Ecol 59: 255–264.
Amoroso A, Domine F, Esposito G, Morin S, Savarino J, Nardino M et al (2010). Microorganisms in dry polar snow are involved in the exchanges of reactive nitrogen species with the atmosphere. Environ Sci Tech 44: 714–719.
Anderson MJ . (2001). A new method for non-parametric multivariate analysis of variance. Austral Ecol 26: 32–46.
Anderson MJ, Willis TJ . (2003). Canonical analysis of principal coordinates: a uselful method of constrained ordination for ecology. Ecology 84: 511–525.
Berry D, Ben Mahfoudh K, Wagner M, Loy A . (2011). Barcoded primers used in multiplex amplicon pyrosequencing bias amplification. Appl Environ Microbiol 78: 612.
Boyd ES, Lange RK, Mitchell AC, Havig JR, Hamilton TL, Lafreniere MJ et al (2011). Diversity, abundance, and potential activity of nitrifying and nitrate-reducing microbial assemblages in a subglacial ecosystem. Appl Environ Microbiol 77: 4778–4787.
Brankatschk R, Towe S, Kleineidam K, Schloter M, Zeyer J . (2011). Abundances and potential activities of nitrogen cycling microbial communities along a chronosequence of a glacier forefield. ISME J 5: 1025–1037.
Brimblecombe P, Tranter M, Abrahams PW, Blackwood I, Davies TD, Vincent CE . (1985). Relocation and preferential elution of acidic solute through the snowpack of a small, remote, high-altitude Scottish catchment. Ann Glaciol 7: 141–147.
Cameron K, Hodson A, Osborn AM . (2012). Carbon and nitrogen biogeochemical cycling potentials of supraglacial cryoconite communities. Polar Biol 35: 1375–1393.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Carpenter EJ, Lin SJ, Capone DG . (2000). Bacterial activity in South Pole snow. Appl Environ Microbiol 66: 4514–4517.
Chuvochina M, Alekhina I, Normand P, Petit J, Bulat S . (2011). Three events of Saharan dust deposition on the Mont Blanc glacier associated with different snow-colonizing bacterial phylotypes. Microbiology 80: 125–131.
Cragin JH, Hewitt AD, Colbeck SC . (1996). Grain-scale mechanisms influencing the elution of ions from snow. Atmos Environ 30: 119–127.
Darcy JL, Lynch RC, King AJ, Robeson MS, Schmidt SK . (2011). Global distribution of Polaromonas phylotypes—evidence for a highly successful dispersal capacity. PLoS One 6: e23742.
Döscher A, Gäggeler HW, Schotterer U, Schwikowski M . (1996). A historical record of ammonium concentrations from a glacier in the Alps. Geophys Res Letts 23: 2741–2744.
Edwards A, Anesio AM, Rassner SM, Sattler B, Hubbard BP, Perkins WT et al (2011). Possible interactions between bacterial diversity, microbial activity and supraglacial hydrology of cryoconite holes in Svalbard. ISME J 5: 150–160.
Erisman JW, Sutton MA, Galloway J, Klimont Z, Winiwarter W . (2008). How a century of ammonia synthesis changed the world. Nat Geosci 1: 636–639.
Etzelmüller B, Ødegård RS, Vatne G, Mysterud RS, Tonning T, Sollid JL . (2000). Glacier characteristics and sediment transfer system of Longyearbreen and Larsbreen, western Spitsbergen. Norweg J Geogr 54: 157–168.
Fierer N, Bradford MA, Jackson RB . (2007). Toward an ecological classification of soil bacteria. Ecology 88: 1354–1364.
Fischer H, Wagenbach D, Kipfstuhl J . (1998). Sulfate and nitrate firn concentrations on the Greenland ice sheet: 2. Temporal anthropogenic deposition changes. J Geophys Res Atmos 103: 21935–21942.
Frey B, Rieder SR, Brunner I, Plötze M, Koetzsch S, Lapanje A et al (2010). Weathering-Associated Bacteria from the Damma Glacier Forefield: Physiological Capabilities and Impact on Granite Dissolution. Appl Environ Microbiol 76: 4788–4796.
Gjessing Y . (1977). Episodic variations of snow concentration of an Arctic snowfield. Atmos Environ 11: 643–647.
Harding T, Jungblut AD, Lovejoy C, Vincent WF . (2011). Microbes in High Arctic Snow and Implications for the Cold Biosphere. Appl Environ Microbiol 77: 3234–3243.
Hodson A . (2006). Biogeochemistry of snowmelt in an Antarctic glacial ecosystem. Water Resources Res 42: W11406.
Hodson A, Anesio AM, Tranter M, Fountain A, Osborn M, Priscu J et al (2008). Glacial ecosystems. Ecol Monogr 78: 41–67.
Hodson A, Roberts T, Engvall A-C, Holmen K, Mumford P . (2010). Glacier ecosystem response to episodic nitrogen enrichment in Svalbard, European High Arctic. Biogeochemistry 98: 171–184.
Hodson AJ, Mumford PN, Kohler J, Wynn PM . (2005). The High Arctic glacial ecosystem: new insights from nutrient budgets. Biogeochemistry 72: 233–256.
Hoham RW, Duval B . (2001). Microbial ecology of snow and freshwater ice with emphasis on snow algae.. In: Jones HG, Pomeroy JW, Walker DA, Hoham RW, (eds) Snow ecology: An Interdisciplinary Examination of Snow-Covered Ecosystems. Cambridge University Press: Cambridge, UK, pp 168–228.
Irvine-Fynn TDL, Edwards A, Newton S, Langford H, Rassner SM, Telling J et al (2012). Microbial cell budgets of an Arctic glacier surface quantified using flow cytometry. Environ Microbiol 14: 2998–3012.
Johannessen M, Henriksen A . (1978). Chemistry of snow meltwater: changes in concentration during melting. Water Resources Res 14: 615–619.
Jones HG . (1999). The ecology of snow-covered systems: a brief overview of nutrient cycling and life in the cold. Hydrol Process 13: 2135–2147.
Kekonen T, Moore J, Perämäki P, Mulvaney R, Isaksson E, Pohjola V et al (2005). The 800 year long ion record from the Lomonosovfonna (Svalbard) ice core. J Geophys Res Atmos 110: D07304.
Kowalchuk G, Stephen J, De Boer W, Prosser J, Embley T, Woldendorp J . (1997). Analysis of ammonia-oxidizing bacteria of the beta subdivision of the class Proteobacteria in coastal sand dunes by denaturing gradient gel electrophoresis and sequencing of PCR-amplified 16S ribosomal DNA fragments. Appl Environ Microbiol 63: 1489–1497.
Kühnel R, Roberts TJ, Björkman MP, Isaksson E, Aas W, Holmén K et al (2011). 20-year climatology of NO3 and NH4 wet deposition at Ny Ålesund, Svalbard. Advances Meteorol 2011: 10.
Larose C, Berger S, Ferrari C, Navarro E, Dommergue A, Schneider D et al (2010). Microbial sequences retrieved from environmental samples from seasonal arctic snow and meltwater from Svalbard, Norway. Extremophiles 14: 205–212.
Larose C, Dommergue A, Vogel TM . (2013). The dynamic arctic snow pack: an unexplored environment for microbial diversity and activity. Biology 2: 317–330.
Mahmood S, Freitag TE, Prosser JI . (2006). Comparison of PCR primer-based strategies for characterization of ammonia oxidizer communities in environmental samples. FEMS Microbiol Ecol 56: 482–493.
Mattes TE, Alexander AK, Richardson PM, Munk AC, Han CS, Stothard P et al (2008). The genome of Polaromonas sp. strain JS666: insights into the evolution of a hydrocarbon- and xenobiotic-degrading bacterium, and features of relevance to biotechnology. Appl Environ Microbiol 74: 6405–6416.
Mindl B, Anesio AM, Meirer K, Hodson AJ, Laybourn-Parry J, Sommaruga R et al (2007). Factors influencing bacterial dynamics along a transect from supraglacial runoff to proglacial lakes of a high Arctic glacieri. FEMS Microbiol Ecol 59: 307–317.
Miteva V, Sowers T, Brenchley J . (2007). Production of N2O by ammonia oxidizing bacteria at subfreezing temperatures as a model for assessing the N2O anomalies in the Vostok ice core. Geomicrobiol J 24: 451–459.
Miteva VI . (2008). Bacteria in snow and glacier ice. In: Margesin R, Schinner F, Marx J-C, Gerday C, (eds) Psychrophiles: From Biodiversity to Biotechnology. Springer Verlag: Berlin, Heidelberg, Germany, pp 31–50.
Morita RY . (1975). Psychrophilic bacteria. Bacteriol Rev 39: 144–167.
Møller AK, Barkay T, Al-Soud WA, Sørensen SJ, Skov H, Kroer N . (2011). Diversity and characterization of mercury-resistant bacteria in snow, freshwater and sea-ice brine from the High Arctic. FEMS Microbiol Ecol 75: 390–401.
Newton RJ, Jones SE, Eiler A, McMahon KD, Bertilsson S . (2011). A guide to the natural history of freshwater lake bacteria. Microbiol Mol Biol Rev 75: 14–49.
Nghiem SV, Hall DK, Mote TL, Tedesco M, Albert MR, Keegan K et al (2012). The extreme melt across the Greenland ice sheet in 2012. Geophys Res Letts 39: L20502.
Polz MF, Hunt DE, Preheim SP, Weinreich DM . (2006). Patterns and mechanisms of genetic and phenotypic differentiation in marine microbes. Phil Trans R Soc B Biol Sci 361: 2009–2021.
Roberts TJ, Hodson A, Evans CD, Holmen K . (2010). Modelling the impacts of a nitrogen pollution event on the biogeochemistry of an Arctic glacier. Ann Glacio 51: 163–170.
Russell NJ . (1990). Cold adaptation of microorganisms. Phil Trans R Soc B Biol Sci 326: 595–611.
Sattler B, Puxbaum H, Psenner R . (2001). Bacterial growth in supercooled cloud droplets. Geophys Res Letts 28: 239–242.
Segawa T, Miyamoto K, Ushida K, Agata K, Okada N, Kohshima S . (2005). Seasonal change in bacterial flora and biomass in mountain snow from the Tateyama Mountains, Japan, analyzed by 16S rRNA gene sequencing and real-time PCR. Appl Environ Microbiol 71: 123–130.
Simoes JC, Zagorodnov VS . (2001). The record of anthropogenic pollution in snow and ice in Svalbard, Norway. Atmos Environ 35: 403–413.
Takeuchi N . (2001). The altitudinal distribution of snow algae on an Alaska glacier (Gulkana Glacier in the Alaska Range). Hydrol Process 15: 3447–3459.
Telling J, Anesio AM, Tranter M, Irvine-Fynn T, Hodson A, Butler C et al (2011). Nitrogen fixation on Arctic glaciers, Svalbard. J Geophys Res 116: G03039.
ter Braak C . (1986). Canonical correspondence analysis: a new eigenvector technique for multivariate direct gradient analysis. Ecology 67: 1167–1179.
Williams MW, Brooks PD, Mosier A, Tonnessen KA . (1996). Mineral nitrogen transformations in and under seasonal snow in a high-elevation catchment in the Rocky Mountains, United States. Water Resources Res 32: 3161–3171.
Xiang S-R, Shang T-C, Chen Y, Jing ZF, Yao T . (2009a). Changes in diversity and biomass of bacteria along a shallow snow pit from Kuytun 51 Glacier, Tianshan Mountains, China. J Geophys Res 114: G04008.
Xiang S-R, Shang T-C, Chen Y, Yao T-D . (2009b). Deposition and postdeposition mechanisms as possible drivers of microbial population variability in glacier ice. FEMS Microbiol Ecol 70: 165–176.
Yagi JM, Sims D, Brettin T, Bruce D, Madsen EL . (2009). The genome of Polaromonas naphthalenivorans strain CJ2, isolated from coal tar-contaminated sediment, reveals physiological and metabolic versatility and evolution through extensive horizontal gene transfer. Environ Microbiol 11: 2253–2270.
Acknowledgements
KH and JZ acknowledge the Austrian Society of Polar Research (ÖGP) for support through the Julius Payer grant and NSINK (Marie Curie Training Action) facilitating the accomplishment of sampling and analyses. We acknowledged Innsbruck University for a scholarship for travel and supply expenses. AE acknowledges the Society for General Microbiology (PFRV 10/4) and Aberystwyth University Research Fund (AU 2010), which supported the completion of this work. We thank Professor C Jamie Newbold, IBERS Aberystwyth for his support of 454 sequencing efforts, and Dr Gareth Wyn Griffith and Dr Tristram Irvine-Fynn for insightful comments on the manuscript. Finally, we are most grateful for the comments of two anonymous referees, which greatly strengthened this version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Hell, K., Edwards, A., Zarsky, J. et al. The dynamic bacterial communities of a melting High Arctic glacier snowpack. ISME J 7, 1814–1826 (2013). https://doi.org/10.1038/ismej.2013.51
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.51
Keywords
This article is cited by
-
Community succession and drivers of CO2-fixing microbes in recently deglaciated soils on the Tibetan Plateau
Journal of Soils and Sediments (2023)
-
Diversity, distribution, and function of bacteria in the supraglacial region hit by glacial lake outburst flood in northern Pakistan
Environmental Sciences Europe (2022)
-
Microbiota entrapped in recently-formed ice: Paradana Ice Cave, Slovenia
Scientific Reports (2021)
-
Exploration of Microbial Diversity of Himalayan Glacier Moraine Soil Using 16S Amplicon Sequencing and Phospholipid Fatty Acid Analysis Approaches
Current Microbiology (2021)
-
Disparity in soil bacterial community succession along a short time-scale deglaciation chronosequence on the Tibetan Plateau
Soil Ecology Letters (2020)
