
Abstract
Human gut microbiota shows high inter-subject variations, but the actual spatial distribution and co-occurrence patterns of gut mucosa microbiota that occur within a healthy human instestinal tract remain poorly understood. In this study, we illustrated a model of this mucosa bacterial communities’ biogeography, based on the largest data set so far, obtained via 454-pyrosequencing of bacterial 16S rDNAs associated with 77 matched biopsy tissue samples taken from terminal ileum, ileocecal valve, ascending colon, transverse colon, descending colon, sigmoid colon and rectum of 11 healthy adult subjects. Borrowing from macro-ecology, we used both Taylor’s power law analysis and phylogeny-based beta-diversity metrics to uncover a highly heterogeneous distribution pattern of mucosa microbial inhabitants along the length of the intestinal tract. We then developed a spatial dispersion model with an R-squared value greater than 0.950 to map out the gut mucosa-associated flora’s non-linear spatial distribution pattern for 51.60% of the 188 most abundant gut bacterial species. Furthermore, spatial co-occurring network analysis of mucosa microbial inhabitants together with occupancy (that is habitat generalists, specialists and opportunist) analyses implies that ecological relationships (both oppositional and symbiotic) between mucosa microbial inhabitants may be important contributors to the observed spatial heterogeneity of mucosa microbiota along the human intestine and may even potentially be associated with mutual cooperation within and functional stability of the gut ecosystem.
Similar content being viewed by others

Introduction
Biogeographic surveys of the human microbiome (Costello et al., 2009) have greatly contributed to our understanding of the role that bacterial communities play in human physiology, nutrition and immunity. (Kau et al., 2011). Among these microbiomes, the human intestinal tract harbors the most abundant, and one of the most diverse, microbial communities known in the human body (Costello et al., 2009; Zhou et al., 2013). The general diversity of the gut microbiome is matched by the observed high level of variance in its composition between individuals (Turnbaugh et al., 2009; Yatsunenko et al., 2012; Schloissnig et al., 2013). Similarly, significant latitudinal variation between surface-adherent and luminal microbial populations has been confirmed by both DNA fingerprinting method (Zoetendal et al., 2002) and polymerase chain reaction-based clone library sequencing, (Eckburg et al., 2005) and plenty of other studies found longitudinal variations of microbial components along the length of the intestinal tract based on cultivation approaches, (Moore and Holdeman, 1974; Hayashi et al., 2002) molecular fingerprinting methods, (Zoetendal et al., 2002; Hayashi et al., 2005; de Carcer et al., 2011) as well as polymerase chain reaction-based clone library sequencing analyses (Hayashi et al., 2002; Hold et al., 2002; Wang et al., 2003, 2005; Eckburg et al., 2005; Frank et al., 2007). Despite a rather well-rounded view of the gut microbiome taken from many viewpoints, to date there has been no quantitative illustration or feasible explanations that help explain the observed longitudinal variations in the intestine, largely due to a dearth of large-scale data. This dilemma prompted us to consider two successive questions in the present study: (1) is there discernible heterogeneity along the intestinal tract; (2) if so, what would modulate the resulting spatial distribution?
In this work, we aimed to perform a comprehensive assessment of spatial patterning of mucosa-associated microbial flora along the intestine tract in healthy human individuals. To this end, we used high-throughput 454-pyrosequencing to examine the bacterial diversity in 11 different healthy subjects’ gut microbiomes by examining 77 mucosa tissue samples, totally composed of seven samples from healthy-appearing sites in each subject: terminal ileum (TI), ileocecal valve (IV), ascending colon (AC), transverse colon (TC), descending colon (DC), sigmoid colon (SC), and rectum (R).
Materials and methods
Subject selection and sampling protocol
The use of human subjects was approved by the Medical Ethics Board of the First People's Hospital of Yunnan Province of China, and all participants provided signed informed written consent prior to being included in this study. In total, there were 11 volunteers (7 females and 4 males), all of whom were unrelated individuals of both sexes who lived in Kunming, China, aged 20–60, who were recruited over four consecutive days in 2010 (detailed information in Supplementary Table S1). The health status of the volunteers was self-reported and confirmed by colonoscopy. No volunteers indicated they had suffered any diseases of the gastrointestinal tract and none had been subjected to surgical procedures for several years prior to this study. No subjects were taking medications at the time of their endoscopy nor had used antibiotics during the year prior to specimen collection.
The colonic mucosa of all 11 subjects appeared grossly normal under endoscope. The morning before colonoscopy, all subjects underwent standard bowel cleansing preparation by drinking 60 ml of a 50% magnesium sulfate solution followed by 2000 ml of water within 1 h. During colonoscopy, seven intestinal mucosal tissue biopsies of approximately 1 × 2 mm each of all 11 subjects were collected from several healthy sites throughout the gut: the terminal ileum (about 155 cm from the anus), ileocecal valve (about 150 cm from the anus), ascending colon (about 142 cm from the anus), transverse colon (about 109 cm from the anus), descending colon (about 64 cm from the anus), sigmoid colon (about 20 cm from the anus) and rectum (about 10 cm from the anus). All tissue biopsies were placed in cryovials without preservative, immediately snap frozen in liquid nitrogen and then stored at –70 °C until transportation on dry ice to Kunming Institute of Zoology, Chinese Academy of Sciences, for sequencing analysis and storage. All samples were stored in their original tubes at –80 °C until further processing. DNA was extracted from biopsy tissue samples, and the V1-V2 region of the 16 S ribosomal RNA gene was amplified, sequenced and analyzed as described in Supplementary Materials and methods.
Taylor’s power law analysis for assessing and interpreting the ‘aggregation degree’ (heterogeneity level) of the human intestinal microbial species and communities
Taylor (1961) discovered that the spatial distribution of many species of various organisms from viruses, bacteria and insects to human populations conforms to a power function relationship:

where m is the population abundance of a single species (a variable largely analogous to population density of a single species) at some spatial sampling site, and V is the corresponding variance. The parameter b, which has been the target of numerous field experiment studies and theoretical analyses, is considered a species-specific dependent on the interaction between species’ behavior and environment (Taylor, 1961, 1984, 2007; Taylor and Taylor, 1977). Theoretical analysis has shown that the power law can be derived from species interactions (Kilpatrick and Ives, 2003; Drossel et al., 2004). The parameter a is mostly influenced by the sampling scheme and is of limited biological significance. Equation (1) can then be transformed into a linear model with the form:

which is often used to estimate the values of parameters a and b. The power function has been verified in numerous field studies, with the model itself being known in ecological studies as Taylor’s power law. The original Taylor’s power law was advanced and applied in macro population ecology, but its extension and novel application to new areas, such as community ecology (multiple species), was recently considered by Ma (2012).
Taylor (1961, 1984) devised a set of rules to determine the types of spatial distribution based on the value of parameter b. When b>1, spatial distribution is aggregated (also termed as contagious); when b=1, the distribution is random, and when b<1, the spatial distribution is regular or uniform. The term ‘distribution’ is used in both the biological and statistical senses. The aggregated (spatial) distribution (sensu biology) generally needs highly skewed statistical distributions such as a Negative Binomial, Neyman’s type A, Polya and Ades distributions to be described mathematically. Random distributions (sensu biology) correspond to Poisson statistical distribution (sensu statistics), whereas regular or uniform (sensu biology) correspond to the uniform distribution (sensu statistics) in statistics. Both random and uniform distributions—though especially uniform—are extremely rare in field populations, even though they are meaningful theoretically. One obvious biological interpretation of the parameter b is that it actually measures the degree of aggregation (or its opposite, dispersion): the larger the b-value, the higher the aggregation degree or the lower the dispersion degree.
In the present study, we estimated the parameter b of Taylor’s power law model (equation (2)) by respectively computing V and m at the microbial community and species level: (i) For individual (subject) independent analysis of mucosal microbial communities (consisting of the n species with at least 10−3 relative abundances), V and m of each species were computed by averaging its abundances of all seven sites, and total n pairs of V and m (equation (2)) per subject were computed for the n species (Table 1), which were used to estimate the parameter b of the power law model for the mucosal microbial community for each individual subject; (ii) For the analysis of mucosal microbial communities for all 11 subjects, V and m of each species were computed by averaging the abundances across 11 subjects. 188 pairs of V and m (equation (2)) per site were computed for the 188 bacterial species, which were used to estimate the power law parameter b of the mucosal microbial community per site (Supplementary Table S3); (iii) For the analysis of single mucosal microbial species of the 11 combined subjects, V and m (equation (2)) of the species’ abundance at each site were computed by averaging the abundances of the 11 subjects, and then seven pairs of V and m per species at each location were computed to determine relative abundances of seven sites (Supplementary Table S4), which were then in turn used to estimate the power law parameter b of the power law model of single mucosal bacterial species.
Community relatedness and phylogenetic analysis
We first applied non-phylogenetic beta diversity metrics (binary jaccard distance) and phylogenetic beta diversity metrics (unweighted UniFrac full tree) (Lozupone and Knight, 2005; Lozupone et al., 2006) to analyze operational taxonomic units’ (OTU) abundance and taxonomic data in order to obtain principal coordinate analysis coordinates needed to compare the intestinal mucosa-associated microbial communities from all 77 samples. To establish the degree to which the sampled gut microbiota was similar in terms of the various compositions of their constituent microbes, we used the method proposed by Ochman et al. (2010) to construct the phylogenies of the 77 samples based on the frequencies of taxonomically assigned OTUs in their gut microbial communities. Character matrices based on all reads were converted into phylogenetic trees using a parsimony-based approach. Each character corresponds to a taxonomically assigned OTU whose frequency in each sample was normalized by coding with one of the four ordered states, reflecting log-unit differences in its occurrence, with an OTU absent from a sample coded as state 0. The range in the occurrence of each OTU across samples (from 0 to 1 400 at the species level) resulted in a five-state data matrix. The matrix was then processed with a heuristic maximum-parsimony-tree search algorithm implemented in PAUP 4.0b10. The default settings of PAUP were used to conduct the tree search.
Single-species abundance-distance dispersion (ADD) model for gut bacteria
Drawing on macro-ecological population dispersal models, such as those studied by Taylor et al. (1979) we propose the following ‘half-log cubic’ model to describe the abundance-distance dispersion (ADD) relationship of bacterial species in the human gut:

We identified the above model through a trial-and-error approach that compared some of the best-performing dispersion models documented in one of Taylor’s earlier studies (Taylor et al., 1979). This model can be fitted to single-species ADD data: single species abundance data of an individual subject as the analysis unit. In this case, X would be the distance from the sampling location to the anus while Y is the abundance of a given OTU in an individual subject. A general criterion of an R-squared value greater than 0.500 was used to determine whether the ADD model would be appropriate for describing single-species ADD.
Co-occurrence network analyses of the human gut microbiome along the length of intestinal tract
We next calculated Pearson’s correlations or associations between the most abundant 188 OTUs (species) along the length of intestinal tract. Each OTU’s abundance was calculated using the mean abundance from all 11 individuals, normalized by log-transformation. Statistic P-values were corrected using the FDR method of the p.adjust package in R. A total of 396 pairs of correlations (278 positive correlations and 118 negative correlations) were found to have an absolute Pearson’s correlation above 0.81 with an FDR-corrected significance level under 0.05, and these correlations were transformed into links between two OTUs in the OTU co-occurrence network. The co-occurrence networks were then visualized using Cytoscape 2.8.2 with a force-directed algorithm (Smoot et al., 2011), and network topological parameters were computed using NetworkAnalyzer (Assenov et al., 2008). Highly connected microbial clusters (modules) in the network were identified using Network Module identification (NeMo) (Rivera et al., 2010) within Cytoscape. The mining of high-confidence modules was largely dependent on both high scores gained by NeMo and high clustering coefficient within modules (clustering coefficients closer to 1.0 represent higher fidelity, with the highest being 1.0).
Results
Summary of pyrosequencing data and spatial change of mucosa microbiota along the length of intestinal tract
Our samples yielded a raw dataset consisting of 270 994 high-quality 16 S ribosomal RNA gene sequences. Using the conventional criterion of 97% sequence similarity at the species level, (Turnbaugh et al., 2010) we identified 7680 OTUs. We next removed 1.11% of the total OTUs that failed to be classified into the bacteria domain with the RDP classifier, leaving 7595 bacterial OTUs. Singletons—a sequence that only occurs once in all 77 samples—were next removed, representing a total of 2510 OTUs. A total of 5085 bacteria OTUs (98.76% of total sequences: 267 632) (Supplementary Table S5) remained, with an average of 649±23 OTUs [Good’s Coverage (estimating what percent of the total species is represented in a sampling site) (Good 1953, Kemp and Aller, 2004): 88.80±0.68%] and 3476±185 sequences per sample (S.E.) (n=77). By performing shared OTU analyses across each of the seven sampling sites for each of the 11 subjects, (Supplementary Table S6) we found that the percentages of shared OTUs (between at least any two sites) ranged from 54.90% to 98.40% (81.60±0.97%), while 18.40±0.97% OTUs were unique to each site. No significance of sequence numbers among all seven sampling sites through the human intestine (Supplementary Table S5) indicated a minimal sequencing bias across any of the seven sampling sites. In addition, no noted significance of Good’s Coverage between all seven sampling sites (Supplementary Table S5) indicated that each sampling site had similar coverage of total microbial species. Accordingly, the relative abundance of OTU may likely be comparable across all seven sampling sites within an individual.
On the basis of the resultant 5085 OTUs, we observed the fluctuation of mucosa microbial components along the length of intestinal tract under phylum, genus and species levels. First, along the intestine (Figure 1c), we found no significant difference of either dominant (e.g., Firmicutes and Bacteroidetes) or rare bacterial phyla (Proteobacteria and Fusobacteria) (Figure 1a). Similarly, three dominant bacterial genera (e.g., Bacteroides, Prevotella and Faecalibacterium) only showed weak variations without significant differences among any of the tested sites (Figure 1b). These results indicated that at a higher taxonomic level, mucosa microbial components potentially tend to be far more stabilized along the intestine. To further visualize spatial variation of mucosa microbiota species within individuals, we used a heatmap-based analysis (Figure 2) on the 188 gut species (from the filtered 5085 OTUs) that had a relative abundance greater than 10−3 (that is, the read number of an OTU divided by total reads, Supplementary Table S2). Of these, there were 79 OTUs (42.02%) shared by at least six subjects (four OTUs of these were by all 11 subjects), 102 OTUs (54.26%) shared by two to five subjects and seven subject-specific OTUs (3.72%). The visualized results (Figure 2) indicated that there may be micro-heterogeneities of mucosa microbial species existing along the intestine within specific individuals, although significant differences across sites were not detected by ANOVA, possibly due to the masking of a high level of inter-subject variation.

Spatial distribution of microbial composition along the length of intestinal tract. (a) bacteria phyla (mean±S.E.); (b) the three most abundant bacterial genera (mean±S.E.); (c) Graphic illustration of seven sites in the gut that appear healthy.

Heat-map showing spatial distributions of 188 bacterial OTUs (species) with at least 10−3 relative abundance per subject. Individual cells are color-coded according to Z-scores to show the normalized abundance of a particular OTU in one site relative to the mean abundance across all seven sites. The relative intensity of the colors indicates how many standard deviations the observed OTU abundance is above or below the mean. All 188 OTUs belong to seven bacterial phyla, mapped left of the heatmap from top to bottom, including Firmicutes, Actinobacteria, Deinococcus-Thermus, Proteobacteria, TM, Fusobacteria and Bacteroidetes. TI, terminal ileum; IV, ileocecal valve; AC, ascending colon; TC, transverse colon; DC, descending colon; SC, sigmoid colon; R, rectum. Note: Heat-map was generated using R.
Taylor’s power law analysis and phylogenetic beta-diversity metrics of mucosa microbiota along the length of intestinal tract
To avoid the potential impact of inter-subject variations on the analysis of inter-site variations, we quantitatively assessed the spatial heterogeneity of mucosa microbiota across all seven sampling sites within individuals. Subsequently, we opted to use Taylor’s power law analysis on gut bacterial community per subject, consisting of the gut species (from the filtered 5085 OTUs) with at least 10−3 relative abundance (that is, the read number of an OTU divided by total reads per subject) (Table 1) (See Materials and methods). We found gut bacterial communities for all 11 subjects had b-values>1 (1.858±0.049) with statistical significance (P<0.0001) as well as a high correlation coefficient (R2=0.754±0.031) (Table 1). The biological interpretation of the parameter b indicates that aggregation degree values greater than 1 denote a high heterogeneity in the spatial distributions of mucosa-associated bacterial community along the intestine within individuals. However, we should caution that in future research it may be necessary to use large-scale 16S rDNA molecular data with high species coverage (that is, high Good’s Coverage of 88.80±0.68% per sampling site in this study) if species abundance—as an indicator of population density of species—is applied to Taylor’s power law analysis. Additionally, we also found that the assessment of the parameter b in Taylor’s power law analysis was independent of using absolute/relative taxonomic abundances as well as sequence differences among seven sampling sites within a single subject (Supplementary Tables S7 and S8). Similarly, test results based on individual-independent phylogenetic beta-diversity metrics (Supplementary Table S9) also suggested that significant differences (Bonferroni corrected P⩽0.01) in community diversities exist among seven sampling sites per subject. In total, based on quantitative assessments from both Taylor’s power analysis and phylogenetic beta-diversity metrics, we consistently found clear, significant heterogeneity of mucosa microbiota along the axis of the intestinal tract within individuals, without any masks from inter-subject variation.
To further verify our preliminary findings, we investigated spatial heterogeneity at the community level based on the combined samples from all 11 subjects, using Taylor’s power law analysis on gut bacterial community consisting of the 188 gut species (Supplementary Table S2) (See Materials and methods). According to the earlier explanation of the parameter b, the power law analysis at the community level (b=1.975±0.021 with both P<0.001 and R2=0.894±0.009) (Supplementary Table S3) revealed non-random or highly aggregated distributions among the gut bacterial community. This finding was independent of different taxonomic levels (phylum, class, order, family, genus and species) (Supplementary Table S3). The consistent pattern was further confirmed using the filtered 5085 OTUs to compare community diversities among seven locations using three phylogenetic beta diversity metrics (Supplementary Materials and methods). The results suggested that significant differences in community diversities (Bonferroni corrected P⩽0.01) exist across all seven sampling sites, though a few exceptions were found after correction of multiple testing (the combined 11 subjects in Supplementary Table S10). Furthermore, we investigated spatial heterogeneity of mucosa microbial species along the intestine based on the combined samples of the 11 subjects, using the 188 gut species (OTUs) (Supplementary Table S4) for Taylor’s power law analysis (See Materials and methods). Our analyzed results from each of the concerned 188 species yielded b-values ranging from 0.5393 to 5.3701 (2.2565±0.0474) (Supplementary Table S4). Totally, 171 of the 188 OTUs (90.96%) had b-values>1 (Supplementary Table S4) with both an accompanying statistical significance (P<0.05 corrected by FDR) and high correlation coefficient (R2=0.878±0.008), demonstrating high heterogeneity in the spatial distribution of single gut species.
Collectively, all our analyzed results from the combined 11 subjects were in concordance with the initial individual-independent findings, together suggesting that there is a distinctive spatial heterogeneity of mucosa-associated microbial community and species along the length of intestinal tract.
Sample clustering based on the difference of mucosa-associated bacteria community
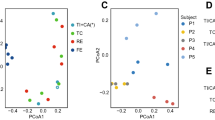
To address the question of what determines the spatial distribution of the human intestinal flora, either potentially local environment selection (site-specific) or historical exposures (individual-specific), we analyzed the relatedness among 77 mucosa-associated bacterial communities using principal coordinate analyses based on non-phylogenetic beta diversity metrics (binary jaccard distance) (Figure 3a) and phylogenetic beta diversity metrics (unweighted UniFrac full tree) (Figure 3b). (Lozupone and Knight, 2005; Lozupone et al., 2006). The results suggest that all 77 samples were partially divided into individual-based groups, such as subjects S407 and S410 (Figures 3a and b). To better discern the relatedness among 77 mucosa-associated bacterial communities, we used a cluster method developed by Ochman et al. (2010) to perform a community relatedness analysis. The results with high bootstrap supports reveal that all 77 samples (Figure 3c) were primarily grouped by individuals with the exception of several samples (e.g., S401). The analysis from a data set of the three American individuals reported by Eckburg et al. (2005) also showed a consistent pattern (Supplementary Figure S1). These findings together suggest that spatial heterogeneity of mucosa microbiota through the intestine might be individual-independent, even when considering the substantial interpersonal variations we noted earlier.

Community relatedness of human intestinal mucosa-associated microbiota. PCoA analyses were used to compare differences between mucosa-associated bacterial communities found in subjects based on non-phylogenetic beta diversity metrics (binary jaccard distance) (a) and phylogenetic beta diversity metrics (unweighted UniFrac full tree) (b) with 100 Monte Carlo randomizations. Each point corresponds to a sample colored by subjects. C, refers to the cluster analysis of 1 000 bootstrap heuristic searches according to parsimonious criterion, which uses the normalized OTU frequency data set with five ordered character states (0, 1, 2, 3 and 4). Branches were colored by the color of the representative individual of the cluster. TI, terminal ileum; IV, ileocecal valve; AC, ascending colon; TC, transverse colon; DC, descending colon; SC, sigmoid colon; R, rectum.
Modeling of spatial dispersion of mucosa-associated microbiota along the intestinal tract
To better understand spatial dispersion of mucosa microbiota along the intestine, we analyzed the ADD relationships of the relevant 188 species for each of the 11 subjects. By performing the ADD analysis with each subject with an R-squared value greater than 0.500, our results illustrated the number of species that fit our ‘half-log cubic’ model in each subject, ranging from 31.40% to 88.52% (57.20±6.34%), and the ADD relationships in 172 of the total 188 species (91.49%) fit the ADD model (Figure 4):

Summary of ADD modeling results. Within the inner line cycle, all 188 species from 11 subjects were used to analyze the abundance-distance dispersion (ADD) relationships of gut microbiota. The number in parentheses indicates that the ADD relationships in 172 of the total 188 species (91.49%) fit the ADD model (a, species at least shared by six subjects; b, species shared by 2 to 5 subjects; c, species unique to subject). Within the middle dashed cycle, the number in parentheses represents how many species in all 188 species existed in each subject (from S400 to S410). Within the outer dashed cycle, the bold number indicates how many species fit the ADD model in each subject (relative percentage was shown in parentheses); X/Y/Z in parentheses: X represents the ADD model fitted species shared by all seven gut sites, Y represents those shared by two to six gut sites and Z represents those unique to specific gut sites.

where x is the distance from sampling location to anus, Y is species abundance and a-d are model parameters. For each subject, the ADD model can describe spatial dispersions of those species shared by all seven sites (e.g., 11/27 in subject S400), those shared by two to six sites (e.g., 16/27 in subject S400) and those unique to one site (e.g., 2/50 in subject S409) (Figure 4). To increase the power of ADD modeling, using more strict R-squared values (0.600, 0.700, 0.800, 0.900 and 0.950, respectively), we found that the ADD relationships in 165, 159, 152, 118 and 97 of the total 188 species (87.77%, 84.57%, 80.85%, 62.77% and 51.60%) fit the ADD model. Totally, our results demonstrated that both spatial dispersion of mucosa microbial inhabitants and their co-occurrence associations through the human intestine could be nonlinear. The biological significance of this described non-linear relationship, though provocative, remains unknown. A similar study suggested that the non-linear relationship could raise a potential spatial segregation of mucosal microbial populations of similar and competing species, and there is a possibility that this spatial segregation could act to stabilize the coexistence of similar species, relaxing the expected interspecific competition (Shigesada et al., 1979).
Co-occurrence network of mucosa-associated microbiota along the intestine
By analyzing and then visualizing the spatial Pearson’s associations or correlations between the 188 gut bacterial species distributed across all seven sampling sites along the human intestine (See Materials and methods), we identified a spatial co-occurrence network of 165 from 188 gut bacterial species with 278 positive correlations (that is, symbiotic relationships between species) and 118 negative correlations (that is, oppositional relationships or colonization resistance between species) (Figure 5a). In the network, (left panel in Figure 5a) four hub species were identified as having the most linkers (14 OTUs co-occurring with each hub): OTU191 (100% similarity to Ruminococcus gnavus ATCC 29149), OTU318 (99% similarity to Faecalibacterium prausnitzii A2-165), OTU1369 (99% similarity to Prevotella copri DSM 18205) and OTU1458 (99% similarity to Anoxybacillus flavithermus WK1). Furthermore, to better understand the biological significance of spatial co-occurrence of mucosa microbiota, we defined those OTUs as being ‘habitat generalist, specialist or opportunist’ based on local abundance and occupancy at the seven sites (see more detailed definitions in the legend of Figure 5). These terms are consistent with those used in a recent study using network analysis to explore co-occurrence patterns in soil microbial communities (Barberan et al., 2012).

Co-occurrence networks of mucosa-associated microbiota along the length of intestinal tract. (a) the network is constructed using 396 spatially co-occurring OTU pairs along the intestine, all with an absolute Pearson’s correlation above 0.81 at a 0.05 FDR-corrected significance level. Left panel: Node colors from bright to dark represent degrees from low to high of each node linked by other nodes. The node labels are the OTU codes. The nodes with the most numerous linkers are defined as hub nodes (square): OTU191, OTU318, OTU1369 and OTU1458. Right panel: OTUs are colored by occupancy (that is, generalists, specialists, habitat OTUs between generalists and specialists, as well as opportunists). Habitat generalist OTUs (in red) are defined as locally abundant (at least two sequences) and appearing in all seven sampling sites in at least six subjects. Habitat specialists OTUs (in blue) are defined as locally abundant (at least two sequences) and appearing in any one of seven sites in at least six subjects. Habitat OTUs between generalists and specialists were colored in gold. Habitat opportunists OTUs (in grey) are defined as locally abundant (one sequence) or appearance in any one of seven sites in less than six subjects. IV, ileocecal valve; AC, ascending colon; TC, transverse colon; DC, descending colon; SC, sigmoid colon; R, rectum. (b) two highly connected microbial clusters (modules). OTUs are colored by occupancy (that is, generalists, specialists and habitat OTUs between generalists and specialists). Edges are colored by positive or negative correlations as described in the left panel of (a). Inter-module interaction was implicated by edges with double-headed arrows. (c) spatial abundance distribution of the 11 bacterial OTUs within two modules (Figure b) as well as quantitative assessments (b-values) of their spatial heterogeneities. b>1 indicates a high heterogeneity in the population spatial distribution that signifies departure from random distribution.
Our results clearly demonstrated spatial co-occurrence patterns of mucosa microbiota together with occupancy (right panel in Figure 5a) and identified habitat generalists (34 of 165 OTUs, such as hub bacteria OTU318), specialists (24 of 165 OTUs, such as hub OTU191 and OTU1369) and opportunists (39 of 165 OTUs). Overall, we observed a significant separation in the co-occurring network analysis between generalists (across hub OTU318) and specialists (across hub OTU1369 and OTU1458, respectively), as well as a distinct overlapping (across hub OTU191) in the co-occurring network analysis between generalists and specialists (right panel in Figure 5).
To further explore the biological significance of spatial co-occurrence network of mucosa microbiota together with occupancy, (Figure 5a) we identified two interplay modules (See Materials and methods) with a high degree of confidence (Figure 5b) associated respectively with either OTU318 or OTU191. OTU318-matched module A with the most stabilized topological structure (clustering coefficient=1.0) was modulated largely by generalists (Figure 5b). Within this module, generalist F. prausnitzii (represented by OTU1457 and increasing towards the rectum) was negatively correlated to and much more abundant (P=0.004) than generalist B. coprophilus (the most abundant in the descending colon) (Figure 5c). Meanwhile, OTU191-matched module B with clustering coefficient of 0.639 was involved in both generalists and specialists (Figure 5b). The abundance of R. gnavus represented by specialist OTU191 (the most abundant in colon) was significantly (P⩽0.001) lower than that of its negative cooperator (generalist OTU1449 with 99% similarity to Bacteroides vulgatus PC510) (the most abundant in terminal ileum and rectum) (Figure 5c). The inter-module interplay was modulated by F. prausnitzii represented by two bacterial OTUs (244 and 2109) (Figure 5b).
Discussion
Currently, high inter-individual variations of gut microbiota within humans has been reported by many studies (Eckburg et al., 2005; Costello et al., 2009; Qin et al., 2010; Arumugam et al., 2011; Claesson et al., 2011; Yatsunenko et al., 2012; Schloissnig et al., 2013). Along the length of the human intestinal tract, spatial heterogeneity of mucosa microbiota remains poorly elucidated, despite previously observed spatial variations of mucosa microbiota (Zoetendal et al., 2002; Eckburg et al., 2005; de Carcer et al., 2011; Hong et al., 2011; Nava et al., 2012). The results of this study allowed us to conclude that there are clear, significant spatial heterogeneities existing in the human intestinal mucosa microbiota within a single individual based on a quantitative Taylor’s power law analyses, which is in accordance with classical phylogenetic beta-diversity metrics of mucosa-associated microbial community diversities across seven sampling sites. Spatial heterogeneities of mucosa microbiota along the intestine could be indicative of underlying differences in gut physiology or a host effect on the microbiome (e.g., diet or lifestyle). This result also expands earlier findings of significant heterogeneity between intestinal luminal, mucosal and fecal microbiota (Zoetendal et al., 2002; Eckburg et al., 2005; Hong et al., 2011) to the scope of mucosa microbiota along the intestinal tract. Spatial co-occurrence patterns of mucosa microbiota together with occupancy (that is habitat generalists, specialists and opportunist) presented in this study provides novel insights into understanding the association between spatial heterogeneity of mucosal-associated intestinal microbiota and human health.
The main trend observed in this study was significant spatial heterogeneity of mucosa microbiota along the human intestine within individuals. This finding was consistent with two facets linked with human gut health or disease. The members of the Enterobacteriaceae were the most abundant in the distal regions of the intestine in health subjects (de Carcer et al., 2011). Otherwise, a large increase in Enterobacteriaceae was found potentially associated with the ileocecal form of Crohn’s disease (CD) (Willing et al., 2009, 2010). Given the scope of this study, local environmental drivers that form spatial heterogeneity of mucosa microbiota through the intestine cannot be determined, but the general trend follows the main physicochemical changes known to occur along the axis of the human intestine.
Several earlier studies suggested that lumen content dehydration and pH increase towards the rectum (Bown et al., 1974), as well as highest short-chain fatty acid , lactate and ethanol concentrations in the proximal colon, decreasing distally with a concomitant increase in products of protein fermentation (e.g., ammonia, branched chain fatty acids and phenolic compounds) (Macfarlane et al., 1992). Likewise, a significant trend from high to low short-chain fatty acid concentrations was again found passing distally from cecum to the descending colon, which provides strong evidence for the occurrence of the microbial breakdown of carbohydrates in the human colon (Cummings et al., 1987). Moreover, another review indicated that glycan-degrading phenotypes are distributed across the length of the human intestinal tract (Koropatkin et al., 2012). Genome analysis suggested that Bacteroides vulgatus PC510 represented by OTU1449 (Supplementary Table S4) encodes arylsulfatases, hexosaminidases, fucosidases and a sialidase with putative roles in harvesting host glycans (Cuív et al., 2011). In addition, mucin composition was the most abundant in the rectum and correlated with the composition of different sulfate-reducing bacteria genera along the human colon (Croix et al., 2011).
Recent studies found that the occurrence of CD is always following a decrease in obligate anaerobes of the phylum Firmicutes and an increase in facultative anaerobes, including members of the family Enterobacteriaceae (Willing et al., 2009, 2010; Joossens et al., 2011). The shift of bacterial communities from obligate to facultative anaerobes strongly suggests a disruption in anaerobiosis and points to a potential role for oxygen in intestinal dysbiosis (Rigottier-Gois 2013). Spatial variation of mucosal oxygen concentration was also implicated along the human colon, especially between the cecum and distal regions (Sheridan et al., 1987). The overgrowth of aerotolerant bacteria with ileal CD (Baumgart et al., 2007; Willing et al., 2009, 2010) was frequently found in several studies, wherein they indicated that the functional stability or disturbance of oxygen-microbe interaction through the intestine may be related to local intestinal health or dysbiosis. Recently, Larsson et al. (2012) found that gut microbiota enables inducing different host responses along the length of the gut. Schluter and Foster (2012) also found that the evolution of the gut microbiota mutualism was dependent on host epithelial selection (Schluter and Foster, 2012). These findings together suggest that spatial heterogeneity of mucosa microbiota through the human intestine may have actually resulted from underlying differences in gut physiology or host effect on the microbiome (again, diet and/or lifestyle). More succinctly put, local environment-microbes or host-microbe interaction may be one of the major driving forces in forming the spatial heterogeneity of the mucosa microbiota along the intestinal tract.
Furthermore, exploring the driving force of microbe-microbe interaction in forming spatial heterogeneity of mucosa microbiota is worth underscoring. Our ADD modeling results imply that it is impossible to use classic multivariate analysis methods to describe spatial co-occurrence relationships across mucosa-associated microbial inhabitants. Unlike classic multivariate analysis methods, correlation (co-occurrence) network analysis allows for the interrogation of nonlinear dynamics of mucosa microbiota over space. This concept was leveraged in the present study, wherein we explored co-occurrence patterns of 165 from 188 gut bacteria species together with occupancy (that is, habitat generalists, specialists and opportunists) (Figure 5a). Co-occurrence network analysis of the healthy human gut microbiota revealed a strong niche specialization with most microbial associations occurring within and between body sites, answering some of the interesting questions as to how healthy microbiota remains stable while showing remarkable variability or spatial heterogeneity within individuals (Faust et al., 2012). Consistent with the finding reported by Faust et al. (2012), via co-occurrence network analysis, our results suggest that ecological relationships (both oppositional and symbiotic) between mucosa microbial inhabitants may also be important contributors to spatial heterogeneity of mucosa microbiota along the human intestinal tract.
In molecular interaction networks, groups of densely connected molecules (network modules) frequently have an important biological significance that may not be readily apparent from other perspectives (Girvan and Newman, 2002; Rives and Galitski, 2003; Newman, 2006). Similarly, network module analyses together with the reported occupancy findings in this study revealed two patterns of spatial microbe-microbe interactions with potentially biological implications. First, the colonization resistance between habitat generalists may be closely related to functional stability of the gut ecosystem along the human intestine, such as generalists F. prausnitzii and B. coprophilus (Module A in Figure 5b). F. prausnitzii is considered to be an anti-inflammatory commensal bacterium identified by gut microbiota analysis of CD patients, (Sokol et al., 2008) and a decrease of F. prausnitzii is closely related to gut dysbiosis in patients with CD (Willing et al., 2010; Joossens et al., 2011). Our study found that F. prausnitzii (generalist OTU1457) was negatively correlated to and significantly (P=0.004) much more abundant than B. coprophilus (generalist OTU1517) (Module A in Figures 5b and c), suggesting there may be colonization resistance between F. prausnitzii and B. coprophilus along the length of the intestinal tract. Accordingly, if the turnover of colonization resistance between those two species occurred, the overgrowth of B. coprophilus along the intestine may result in a decrease of F. prausnitzii possibly linked with the presence of gut dysbiosis, such as CD (Willing et al., 2010; Joossens et al., 2011).
Our results suggest that the colonization resistance between habitat generalists and specialists may be associated with local functional stability of the gut ecosystem, such as generalist B. vulgates (OTU1449) and specialist R. gnavus (OTU191) to AC (Module B in Figure 5b). Beneficial bacteria B. vulgatus is amongst the most commonly isolated microbes from the human gastrointestinal tract in healthy humans (Tap et al., 2009; Qin et al., 2010). If this were not the case, Willing et al.’s (2010) study suggested that an overrepresentation of R. gnavus is closely related to the ileocecal form of Dour study; however, we found that B. vulgatus (generalist OTU1449) was negatively correlated to and significantly (P⩽0.001) much more abundant than R. gnavus (specialist OTU191 to AC) (Module B in Figures 5b and c), implying that there may be local colonization resistance between B. vulgatus and R. gnavus in AC, possibly linked with the healthy status of AC in humans. This finding further indicated that an overgrowth of R. gnavus may induce local gut dysbiosis, such as the AC form of CD, similar to the ileocecal CD (Willing et al., 2010). Interestingly, the interplay between the two modules (Figure 5b) indicated that the two patterns that we described were interdependent but not isolated, possibly associated with local gut health. Willing et al. (2010) found that local gut dysbiosis specific to patients with ileal CD included the disappearance of core bacteria, such as Faecalibacterium (corresponding to generalist OTU 318 and OTU1447 within Module A in Figure 5b) and increased amounts of R. gnavus (corresponding to specialist OTU 191 within Module B in Figure 5b) (Willing et al., 2010), partially supporting our inference stated above.
These collected findings together suggest that spatially ecological relationships (both oppositional and symbiotic) between mucosa microbial inhabitants may potentially be associated with local gut health and, by extension, stability of the gut microbiome, though further research is clearly needed to explain the specific role each species actually plays in the intestinal tract.
Conclusion
Using a multidisciplinary approach, this study demonstrated both spatial heterogeneity and co-occurrence pattern of mucosa microbiota together with occupancy along the length of the human intestinal tract. Given the local nature of various gut dysbiosis (e.g., colorectal cancers and inflammatory diseases), further research on how the functionality of mucosa-associated microbiome could potentially change along the human intestine, as well as the nature of local host–microbe interactions will likely strengthen our findings reported in this present study.
Accession codes
References
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR et al (2011). Enterotypes of the human gut microbiome. Nature 473: 174–180.
Assenov Y, Ramírez F, Schelhorn S-E, Lengauer T, Albrecht M . (2008). Computing topological parameters of biological networks. Bioinformatics 24: 282–284.
Barberan A, Bates ST, Casamayor EO, Fierer N . (2012). Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J 6: 343–351.
Baumgart M, Dogan B, Rishniw M, Weitzman G, Bosworth B, Yantiss R et al (2007). Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn's disease involving the ileum. ISME J 1: 403–418.
Bown RL, Gibson JA, Sladen GE, Hicks B, Dawson AM . (1974). Effects of lactulose and other laxatives on ileal and colonic pH as measured by a radiotelemetry device. Gut 15: 999–1004.
Claesson MJ, Cusack S, O'Sullivan O, Greene-Diniz R, de Weerd H, Flannery E et al (2011). Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci USA 108: 4586–4591.
Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R . (2009). Bacterial community variation in human body habitats across space and time. Science 326: 1694–1697.
Croix JA, Carbonero F, Nava GM, Russell M, Greenberg E, Gaskins HR . (2011). On the relationship between sialomucin and sulfomucin expression and hydrogenotrophic microbes in the human colonic mucosa. PLoS One 6: e24447.
Cuív PÓ, Klaassens ES, Durkin AS, Harkins DM, Foster L, McCorrison J et al (2011). Draft genome sequence of Bacteroides vulgatus PC510, a strain isolated from human feces. J Bacteriol 193: 4025–4026.
Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT . (1987). Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 28: 1221–1227.
de Carcer DA, Cuiv PO, Wang T, Kang S, Worthley D, Whitehall V et al (2011). Numerical ecology validates a biogeographical distribution and gender-based effect on mucosa-associated bacteria along the human colon. ISME J 5: 801–809.
Drossel B, McKane AJ, Quince C . (2004). The impact of nonlinear functional responses on the long-term evolution of food web structure. J Theor Biol 229: 539–548.
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M et al (2005). Diversity of the human intestinal microbial flora. Science 308: 1635–1638.
Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J et al (2012). Microbial co-occurrence relationships in the human microbiome. PLoS Comput Biol 8: e1002606.
Frank DN, St. Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR . (2007). Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA 104: 13780–13785.
Good IJ . (1953). The population frequencies of species and the estimation of population parameters. Biometrika 40: 237–264.
Girvan M, Newman MEJ . (2002). Community structure in social and biological networks. Proc Natl Acad Sci USA 99: 7821–7826.
Hayashi H, Sakamoto M, Benno Y . (2002). Phylogenetic analysis of the human gut microbiota using 16S rDNA clone libraries and strictly anaerobic culture-based methods. Microbiol Immunol 46: 535–548.
Hayashi H, Takahashi R, Nishi T, Sakamoto M, Benno Y . (2005). Molecular analysis of jejunal, ileal, caecal and recto-sigmoidal human colonic microbiota using 16S rRNA gene libraries and terminal restriction fragment length polymorphism. J Med Microbiol 54: 1093–1101.
Hold GL, Pryde SE, Russell VJ, Furrie E, Flint HJ . (2002). Assessment of microbial diversity in human colonic samples by 16S rDNA sequence analysis. FEMS Microbiol Ecol 39: 33–39.
Hong PY, Croix JA, Greenberg E, Gaskins HR, Mackie RI . (2011). Pyrosequencing-based analysis of the mucosal microbiota in healthy individuals reveals ubiquitous bacterial groups and micro-heterogeneity. PLoS One 6: e25042.
Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, Rutgeerts P et al (2011). Dysbiosis of the faecal microbiota in patients with Crohn's disease and their unaffected relatives. Gut 60: 631–637.
Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI . (2011). Human nutrition, the gut microbiome and the immune system. Nature 474: 327–336.
Kemp PF, Aller JY . (2004). Bacterial diversity in aquatic and other environments: what 16S rDNA libraries can tell us. FEMS Microbiol Ecol 47: 161–177.
Kilpatrick AM, Ives AR . (2003). Species interactions can explain Taylor's power law for ecological time series. Nature 422: 65–68.
Koropatkin NM, Cameron EA, Martens EC . (2012). How glycan metabolism shapes the human gut microbiota. Nat Rev Micro 10: 323–335.
Larsson E, Tremaroli V, Lee YS, Koren O, Nookaew I, Fricker A et al (2012). Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut 61: 1124–1131.
Lozupone C, Knight R . (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71: 8228–8235.
Lozupone C, Hamady M, Knight R . (2006). UniFrac - An online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 7: 371.
Ma ZS . (2012). http://adsabs.harvard.edu/abs/2012arXiv1205.3504M.
Macfarlane GT, Gibson GR, Cummings JH . (1992). Comparison of fermentation reactions in different regions of the human colon. J Appl Microbiol 72: 57–64.
Moore WE, Holdeman LV . (1974). Human fecal flora: the normal flora of 20 Japanese-Hawaiians. Appl Microbiol 27: 961–979.
Nava GM, Carbonero F, Croix JA, Greenberg E, Gaskins HR . (2012). Abundance and diversity of mucosa-associated hydrogenotrophic microbes in the healthy human colon. ISME J 6: 57–70.
Newman MEJ . (2006). Modularity and community structure in networks. Proc Natl Acad Sci USA 103: 8577–8582.
Ochman H, Worobey M, Kuo C-H, Ndjango J-BN, Peeters M, Hahn BH et al (2010). Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biol 8: e1000546.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C et al (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59–65.
Rigottier-Gois L . (2013). Dysbiosis in inflammatory bowel diseases: the oxygen hypothesis. ISME J 7: 1256–1261.
Rivera C, Vakil R, Bader J . (2010). NeMo: network module identification in Cytoscape. BMC Bioinformatics 11: S61.
Rives AW, Galitski T . (2003). Modular organization of cellular networks. Proc Natl Acad Sci USA 100: 1128–1133.
Schloissnig S, Arumugam M, Sunagawa S, Mitreva M, Tap J, Zhu A et al (2013). Genomic variation landscape of the human gut microbiome. Nature 493: 45–50.
Schluter J, Foster KR . (2012). The evolution of mutualism in gut microbiota via host epithelial selection. PLoS Biol 10: e1001424.
Sheridan W, Lowndes R, Young H . (1987). Tissue oxygen tension as a predictor of colonic anastomotic healing. Dis Colon Rectum 30: 867–871.
Shigesada N, Kawasaki K, Teramoto E . (1979). Spatial segregation of interacting species. J Theor Biol 79: 83–99.
Smoot ME, Ono K, Ruscheinski J, Wang P-L, Ideker T . (2011). Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27: 431–432.
Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux J-J et al (2008). Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA 105: 16731–16736.
Tap J, Mondot S, Levenez F, Pelletier E, Caron C, Furet J-P et al (2009). Towards the human intestinal microbiota phylogenetic core. Environ Microbiol 11: 2574–2584.
Taylor LR . (1961). Aggregation, variance and the mean. Nature 189: 732–735.
Taylor LR . (1984). Assessing and interpreting the spatial distributions of insect populations. Annu Rev Entomol 29: 321–357.
Taylor RAJ . (2007). Obituary: Roy (L. R.) Taylor (1924–2007). J Anim Ecol 76: 630–631.
Taylor LR, Taylor RAJ . (1977). Aggregation, migration and population mechanics. Nature 265: 415–421.
Taylor LR, Woiwod IP, Perry JN . (1979). The negative binomial as a dynamic ecological model for aggregation, and the density dependence of k. J Anim Ecol 48: 289–304.
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE et al (2009). A core gut microbiome in obese and lean twins. Nature 457: 480–484.
Turnbaugh PJ, Quince C, Faith JJ, McHardy AC, Yatsunenko T, Niazi F et al (2010). Organismal, genetic, and transcriptional variation in the deeply sequenced gut microbiomes of identical twins. Proc Natl Acad Sci USA 107: 7503–7508.
Wang M, Ahrné S, Jeppsson B, Molin G . (2005). Comparison of bacterial diversity along the human intestinal tract by direct cloning and sequencing of 16S rRNA genes. FEMS Microbiol Ecol 54: 219–231.
Wang X, Heazlewood SP, Krause DO, Florin THJ . (2003). Molecular characterization of the microbial species that colonize human ileal and colonic mucosa by using 16S rDNA sequence analysis. J Appl Microbiol 95: 508–520.
Willing B, Halfvarson J, Dicksved J, Rosenquist M, Jarnerot G, Engstrand L et al (2009). Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn's disease. Inflamm Bowel Dis 15: 653–660.
Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z et al (2010). A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology 139: 1844–1854.
Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M et al (2012). Human gut microbiome viewed across age and geography. Nature 486: 222–227.
Zhou Y, Gao H, Mihindukulasuriya K, Rosa P, Wylie K, Vishnivetskaya T et al (2013). Biogeography of the ecosystems of the healthy human body. Genome Biol 14: R1.
Zoetendal EG, von Wright A, Vilpponen-Salmela T, Ben-Amor K, Akkermans ADL, de Vos WM . (2002). Mucosa-associated bacteria in the human gastrointestinal tract are uniformly distributed along the colon and differ from the community recovered from feces. Appl Environ Microbiol 68: 3401–3407.
Acknowledgements
The authors would like to thank Kunming Biological Diversity Regional Center of Large Apparatus and Equipments, Kunming Institute of Zoology, Chinese Academy of Sciences for their superb technical assistances as well as Andrew Willden (Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, China) for improving the language of the manuscript. This research was supported by the following grants: National Natural Science Foundation of China (Grant No. 31100916 to ZZ and Grant No. 61175071 to ZM); Natural Science Foundation of Yunnan Province of China (Grant No. 2011FA035 to PS and Grant No. 2010CD191 to JG); ‘A Hundred Talent Program’ from Chinese Academy of Sciences to PS and ZM, respectively; the three grants to ZM (that is, ‘Top Talents Program in Science and Technology’ from Yunnan Province, ‘Top Talents from Overseas’ from Yunan Province and ‘Innovative Research Initiative of the Synergy between the Natural and Computational Evolutions’ of CAS-Yunan Province). Sequences were deposited in the NCBI Sequence Read Archive (accession number SRA052611).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
ZZ, JG and PS designed the experiments. ZZ, JG, XT and HF generated the data. ZZ, JG, JX, XW, ZM and PS analysed the data. ZZ, ZM and PS wrote the manuscript with inputs from the other members of the team. All authors read and approved the final manuscript.
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article
Cite this article
Zhang, Z., Geng, J., Tang, X. et al. Spatial heterogeneity and co-occurrence patterns of human mucosal-associated intestinal microbiota. ISME J 8, 881–893 (2014). https://doi.org/10.1038/ismej.2013.185
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.185
Keywords
This article is cited by
-
Correlation analysis of lung mucosa-colonizing bacteria with clinical features reveals metastasis-associated bacterial community structure in non-small cell lung cancer patients
Respiratory Research (2023)
-
Genetic hypogonadal mouse model reveals niche-specific influence of reproductive axis and sex on intestinal microbial communities
Biology of Sex Differences (2023)
-
Gut microbiota in colorectal cancer development and therapy
Nature Reviews Clinical Oncology (2023)
-
Gut microbiome signatures of extreme environment adaption in Tibetan pig
npj Biofilms and Microbiomes (2023)
-
Identifying keystone species in microbial communities using deep learning
Nature Ecology & Evolution (2023)
