
Abstract
An uncultured member of the phylum Chlorobi, provisionally named ‘Candidatus Thermochlorobacter aerophilum’, occurs in the microbial mats of alkaline siliceous hot springs at the Yellowstone National Park. ‘Ca. T. aerophilum’ was investigated through metagenomic and metatranscriptomic approaches. ‘Ca. T. aerophilum’ is a member of a novel, family-level lineage of Chlorobi, a chlorophototroph that synthesizes type-1 reaction centers and chlorosomes similar to cultivated relatives among the green sulfur bacteria, but is otherwise very different physiologically. ‘Ca. T. aerophilum’ is proposed to be an aerobic photoheterotroph that cannot oxidize sulfur compounds, cannot fix N2, and does not fix CO2 autotrophically. Metagenomic analyses suggest that ‘Ca. T. aerophilum’ depends on other mat organisms for fixed carbon and nitrogen, several amino acids, and other important nutrients. The failure to detect bchU suggests that ‘Ca. T. aerophilum’ synthesizes bacteriochlorophyll (BChl) d, and thus it occupies a different ecological niche than other chlorosome-containing chlorophototrophs in the mat. Transcription profiling throughout a diel cycle revealed distinctive gene expression patterns. Although ‘Ca. T. aerophilum’ probably photoassimilates organic carbon sources and synthesizes most of its cell materials during the day, it mainly transcribes genes for BChl synthesis during late afternoon and early morning, and it synthesizes and assembles its photosynthetic apparatus during the night.
Similar content being viewed by others
Introduction
Until the recent discovery of Ignavibacterium album (Iino et al., 2010), which was initially described as a non-chlorophototrophic, obligately anaerobic, fermentative organism, cultivated representatives from the phylum Chlorobi were a group of physiologically similar and phylogenetically coherent anoxygenic chlorophototrophs that rely on BChl-based phototrophy, that is, chlorophototrophy, as their principal energy source. These organisms included all bacteria commonly known as green sulfur bacteria (GSB; Chlorobiaceae) (Overmann, 2001; Bryant and Frigaard, 2006; Bryant et al., 2012). Cultivated members of the GSB lack swimming motility and are strict anaerobes and obligate chlorophotoautotrophs. With the exception of Chlorobium ferrooxidans (Heising et al., 1999), all characterized GSB can use sulfide, and most can use other reduced sulfur compounds, as an electron donor for CO2 fixation (Overmann, 2008; Frigaard and Dahl, 2009; Gregersen et al., 2011). Other universally distributed and characteristic properties of GSB include their ability to reduce dinitrogen via molybdenum-containing nitrogenase (Wahlund and Madigan, 1993), and their ability to fix CO2 via the reverse TCA cycle (Buchanan and Arnon, 1990; Wahlund and Tabita, 1997; Tang et al., 2011).
Analyses of 16S rRNA sequences of cultured organisms and environmental samples have suggested that cultured representatives of the Chlorobiaceae represent essentially one monophyletic and late-diverging clade of the phylum Chlorobi (Iino et al., 2010). However, the discovery of I. album (Iino et al., 2010), whose genome indicates that it is in fact an aerobic, flagellated, heterotroph (Liu Z et al., submitted for publication), has raised many questions about the physiological diversity of members of the Chlorobi that are distantly related to cultivated GSB strains.
This study describes the properties of a population of organisms discovered in microbial mats associated with Mushroom and Octopus Springs, alkaline siliceous hot springs within the Lower Geyser Basin of Yellowstone National Park, WY, USA (Klatt et al., 2011; Liu et al., 2011). In addition to abundant chlorophototrophic members of the genera Synechococcus and Roseiflexus (Weller et al., 1992), analyses of 16S rRNA sequences revealed the presence of organisms tentatively assigned to GSB (Ferris et al., 1996; Ferris and Ward, 1997). The distribution of 16S rRNA sequences with depth revealed that these organisms were most abundant at a depth of 300–600 μm, where the mats become highly oxygenated under strong illumination, because Synechococcus spp. populations also thrive throughout the upper 0.5–1.0 mm thick photic zone of the mat (Ramsing et al., 2000). These observations are clearly inconsistent with the general perception that GSB are strict anaerobes, which are highly sensitive to oxygen in the light (Li et al., 2009), and they precluded any definitive conclusions about the true identity of the organisms contributing these 16S rRNA sequences. The traditional method to investigate the physiological properties of such organisms would be through cultivation studies with axenic cultures. However, past and ongoing cultivation efforts to isolate such organism(s) have unfortunately been unsuccessful.
Rapidly developing DNA and RNA sequencing technologies provided new approaches to study these organisms. Analyses of metagenomic data from Mushroom and Octopus Springs identified sequences encoding unusual homodimeric type-1 photosynthetic reaction centers. One of the pscA genes belonged to a previously unknown chlorophototroph, ‘Candidatus Chloracidobacterium thermophilum’ (‘Ca. C. thermophilum’), whereas the other belonged to a GSB-like organism, which was tentatively named ‘OS GSB’ (Bryant et al., 2007). Recently, a more complete study of the metagenome showed that seven chlorophototrophic populations, including ‘OS GSB’, Synechococcus Types A and B′, ‘Ca. C. thermophilum’, and three different Chloroflexi populations (Roseiflexus, Chloroflexus and Anaerolineae-like spp.) dominate the chlorophototrophic microbial communities (Klatt et al., 2011). Sequences derived from the different populations were separated into clusters using k-means clustering algorithm that is based on frequency patterns of tri-, tetra-, penta- and hexa-nucleotides among scaffolds larger than 20 kb (Teeling et al., 2004; Klatt et al., 2011). Phylogenetic marker genes and photosynthesis genes from ‘OS GSB’ sequences clearly established a relationship between ‘OS GSB’ and currently cultivated GSB, especially C. thalassium, with nucleotide sequence identity values of ∼63% between ‘OS GSB’ and C. thalassium (Klatt et al., 2011). A metatranscriptomics study of the mat community in Mushroom Spring confirmed the findings of the metagenome study and demonstrated the potential of applying next-generation-sequencing technology to studies of global transcription of the major populations in the microbial communities (Liu et al., 2011). The data obtained from the metagenomic and metatranscriptomic studies are highly complementary. For example, incomplete coverage is a major drawback when metagenomic data are used to infer the physiology of organisms. However, transcripts of physiologically important genes can often be detected in a metatranscriptome, even if the corresponding genes are missing from the metagenome. Furthermore, transcription patterns of genes provide information about the cellular functions of gene products that cannot be inferred from analyzing sequences alone.
In this study, we describe the results of detailed analyses of the metagenome of ‘OS GSB’ and inferences about the physiology of these organisms derived from analyses of the diel expression patterns of the corresponding metatranscriptome. These data suggest that the ‘OS GSB’ organisms are dramatically different from currently described GSB. We suggest that they belong to a new family within the phylum Chlorobi, Thermochlorobacteriaceae, whose members include aerobic, photoheterotrophs that are unable to use sulfide as an electron donor. A model of the cellular physiology of these previously undescribed, moderately thermophilic microorganisms over a diel cycle is proposed, and possible interactions with other chlorophototrophic populations in the mats are discussed.
Materials and methods
Metagenome of ‘OS GSB’
The sequencing and assembly of the metagenome scaffolds of the Mushroom and Octopus Springs chlorophototrophic mat communities and the clustering of scaffolds associated with various bacterial populations were described previously (Klatt et al., 2011). The ‘OS GSB’ metagenome comprises one of the eight clusters of scaffolds (>20 kb), which contained phylogenetically meaningful genes, such as genes for 16S rRNA and ribosomal proteins, and which were characteristic of members of the phylum Chlorobi, especially the family Chlorobiaceae. The metagenome additionally contained other scaffolds (5–20 kb) that contained sequences with similarity to genes found in the genomes of other members of the Chlorobi. The criteria for the identification of these scaffolds were previously described (Liu et al., 2011).
Collection, preparation, sequencing, and analyses of metatranscriptome samples
A total of 24 samples of the microbial mat growing at 60–61 °C were collected on 11 September 2009–12 September 2009 at the start of each hour at Mushroom Spring, Yellowstone National Park, WY, USA as described (Liu et al., 2011). For each time point, RNA was isolated and pooled from the top ∼2-mm layer from two independent #4 core mat samples (∼1 cm2). The irradiance during the collection period was recorded as the downwelling quantum irradiance (400–700 nm; μmol photons m−2 s−1) using a datalogger LI-1400 light meter (LiCor, Lincoln, NE, USA) equipped with a LI-192 irradiance sensor (LiCor). Oxygen concentrations were measured in situ using microelectrodes as described previously (Jensen et al., 2011).
The storage of retrieved microbial mat samples, extraction of RNA, preparation of cDNA and sequencing protocols were the same as described (Liu et al., 2011), except that cDNA was only sequenced using SOLiD technology. Because of a sample-processing error, no data are available for the 11:00 AM time point. Metatranscriptomic data were analyzed as described (Liu et al., 2011) with the modification that mRNA counts for genes were normalized to the total mRNA counts instead of the total rRNA counts for each organism (see Supplementary Information for details).
Results and discussion
Taxonomy of ‘OS GSB’
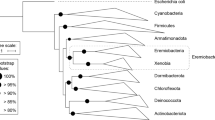
A previous analysis of assembled metagenomic data as well as preliminary metatranscriptomic analyses provided evidence for the existence of organisms distantly related to the GSB Chloroherpeton thalassium in the mats of Octopus and Mushroom Springs (Klatt et al., 2011; Liu et al., 2011).The 16S rRNA sequence recovered from the metagenome scaffolds of ‘OS GSB’ is most similar to sequences of members of Chlorobea (87.7% identity to C. thalassium) among the six class-level lineages of Chlorobi described by (Iino et al., 2010). Figure 1 shows an analysis of 16S rRNA sequences that mainly focuses on the class Chlorobea. ‘OS GSB’ and some other uncultured organisms form a clade that is well separated from the clade that contains the four best-studied genera of GSB: Chlorobium, Chlorobaculum, Prosthecochloris and Chloroherpeton (Imhoff, 2003; Imhoff and Thiel, 2010; Bryant et al., 2012). Four of the five GSB-like 16S rRNA sequence types (E, E′, E″, III-9) previously detected in Octopus spring (Ward et al., 1998) were very similar to ‘OS GSB’ (95–100% identical). Because all sequences in this new clade were obtained from hot-spring environments (Fouke et al., 2003; Lau et al., 2009; Fouke, 2011), we propose to name this novel clade ‘Thermochlorobacteriaceae’. The phylogenetic distances between members of the Thermochlorobacteriaceae and the genera Chlorobium, Chlorobaculum and Prosthecochloris, are much longer than those among the latter three genera, but are obviously shorter than the distances between Thermochlorobacteriaceae and other classes such as Ignavibacteria. Therefore, it seems appropriate to designate the clade ‘Thermochlorobacteriaceae’ as a novel family within the class Chlorobea, order Chlorobiales. These data also suggest that ‘Chloroherpeton’ is actually on the same phylogenetic level as Thermochlorobacteriaceae, and it should therefore be regarded as a family (Chloroherpetonaceae) instead of a genus within the Chlorobiaceae. Similar observations and suggestions were made recently on the basis of preliminary genome sequence comparisons (Bryant et al., 2012).

16S rRNA phylogenetic tree of the order Chlorobiales, class Chlorobea. Neighbor joining tree of 16S rRNA tree of selected Chlorobi sequences. Sequences were obtained from the RDP database (Cole et al., 2009). The tree was generated using MEGA (Tamura et al., 2007) with Jukes-Cantor correction. Very similar or identical sequences were removed to simplify the tree but this did not alter the tree topology. Bootstrap support values based on 1000 bootstrap samplings were shown for each node. Bar denotes 0.02 changes per nucleotide site.
Overview of the metagenome of ‘OS GSB’
The metagenome of ‘OS GSB’ consisted of 80 scaffolds larger than 5 kb, which summed to 3.18 Mb, with a mol G+C% content of 47.5%. This metagenome encoded 2775 predicted protein-coding sequences (CDS). Among these CDS, 147 pairs of genes were potentially redundant copies (genes that were >95% identical to one another in nucleotide sequence). When the redundant genes were removed, the metagenome size and the number of CDS decreased to 3.04 Mb and 2628 CDS, respectively. Both numbers are larger than typical genome sizes among GSB, but they are similar to those of C. thalassium (3.29 Mb and 2710 CDS) (Bryant et al., 2012). Thus, these data suggested that the metagenome of ‘OS GSB’ was nearly complete. The data also suggested that sequences derived from members of this population were mostly assembled into a single consensus sequence, which implied that the individual sequences were very similar. In comparison, sequences of the two main Synechococcus spp. populations in the same community, types A and B′, which are ∼86% identical in nucleotide sequence to each other, were separately assembled into two sets of scaffolds in the same metagenome, which contained the majority of the genes found in two corresponding isolate genomes (Bhaya et al., 2007; Liu et al., 2011). This comparison indicates that ‘OS GSB’ comprises a relatively uniform population and that sequence differences within the population are smaller than those between the Synechococcus spp. type A and B′ genomes. This observation is consistent with previous analyses showing that the single 16S rRNA sequence type for ‘OS GSB’ recruited 100% of the 16S rRNA sequences recovered by pyrosequencing of cDNA (Liu et al., 2011), most of which were >99% identical to ‘OS GSB’ 16S rRNA sequences from the mat (Supplementary Figure 1). Similar results were observed with a set of PCR-amplified, Sanger-sequenced, 16S rRNA sequences from the same mat, which were assigned to the phylum Chlorobi (Supplementary Figure 1). Therefore, on the basis of these observations, this population has provisionally been named ‘Ca. T. aerophilum’.
Figure 2 shows the phylogenetic distribution of the best hits of predicted ‘Ca. T. aerophilum’ proteins in the NCBI nr database. Proteins from GSB, mainly those of C. thalassium, accounted for 59% of the best hits. These data clearly demonstrate the close relationship between ‘Ca. T. aerophilum’ and other GSB. A substantial percentage (11.5%) of the proteins were most similar to proteins found in members of the Bacteriodetes, organisms that share a common ancestry with Chlorobi (Ludwig and Klenk, 2001; Ciccarelli et al., 2006). Interestingly, 1.6% of ‘Ca. T. aerophilum’ proteins were most similar to proteins of ‘Ca. C. thermophilum’, a newly discovered chlorophototrophic member of the phylum Acidobacteria from the same mat community (Bryant et al., 2007; Garcia Costas et al., 2012). This suggests possible horizontal gene transfers may have occurred between ‘Ca. T. aerophilum’ and ‘Ca. C. thermophilum’.

Phylogenetic distribution of best hits of ‘Ca. T. aerophilum’ proteins. Data were obtained based on a BLASTP search of ‘Ca. T. aerophilum’ proteins against the nr database and the complete genome of ‘Ca. C. thermophilum’ (Garcia Costas et al., 2012) with an e-value cutoff of 0.001.
Among a set of 813 orthologous genes from the core genome deduced from 12 individual GSB genomes (Bryant et al., 2012), homologs of 125 genes were not found in the ‘Ca. T. aerophilum’ metagenome (see Supplementary Table 1), some of which probably resulted from the incompleteness of the metagenome. As discussed above, transcripts for physiologically important genes should be relatively abundant in the metatranscriptome. Therefore, the metatranscriptome was searched in attempts to identify transcripts for some of the most important but potentially missing genes (see Supplementary Information for details). Because C. thalassium proteins are most similar in sequence to those of ‘Ca. T. aerophilum’ (Figure 2), whenever possible they were used as queries in the searches. Because of the short length (50 bp) of the cDNA sequences obtained from SOLiD technology, detection of transcripts requires very high sequence conservation. Thus, negative results were only considered to support strongly the probable absence of a gene (and the encoded protein) when homologous proteins in GSB were extremely conserved and/or when transcripts of homologous genes of other, more phylogenetically distantly related organisms, were detected during the same search. The results of these searches are compiled in Table 1, and the implications of these results are discussed below.
Overview of the metatranscriptome of ‘Ca. T. aerophilum’
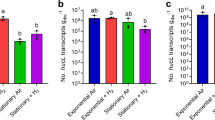
The metatranscriptome of ‘Ca. T. aerophilum’ included from 50 000 to >300 000 mRNA sequences for each timepoint during the diel sampling (Table 2). A total of 2545 genes (96.8%) had at least one aligned sequence, and among these genes, 1818 genes (71%) exhibited a statistically significant difference for at least one pair of time points. These numbers are comparable to those previously observed in a preliminary metatranscription study of this mat community (Liu et al., 2011). When normalized by the total numbers of cDNA sequences for each sample, the numbers of ‘Ca. T. aerophilum’ mRNA sequences were significantly higher during the light period than the dark period (Table 2). These data suggested that the transcriptional activities of this population were highest under strong solar irradiance, that is, during the day, which also coincided with the highest O2 levels (Figure 3).

Examples of clusters of genes based on their in situ diel patterns of relative transcript levels in the microbial mat of Mushroom Spring. The upper panel shows in situ downwelling irradiance (black line) and O2 concentration at the microbial mat surface (red line) and 0.6 mm below the surface (green line). Each column in the heat-map style graph represents normalized transcription levels of genes at a time point. Data for 11:00 AM were missing (see text). Each row represents a gene. Red indicates relatively high transcript levels while green indicates relatively low transcript levels. Degrees of variations are reflected in color intensities.
Patterns of transcript abundance for genes were clustered into three major patterns (see Supplementary Information for clustering methods): a ‘day pattern’, for which transcript level increased and decreased in the same manner as solar irradiance; a ‘night pattern’, for which the exact opposite of the ‘day pattern’ was observed; and an ‘afternoon pattern’, for which transcript abundance was generally higher during the day but was obviously highest in the late afternoon (Figure 3). Genes with the same patterns could be further separated into clusters because of different degrees of variation (e.g. ‘stronger’ or ‘weaker’ day pattern; Figure 3). Reflecting the overall trend described above, most genes were assigned to the ‘day pattern’ cluster. However, in the other two smaller clusters (1.9% and 3.6% of all clustered genes for ‘night pattern’ and ‘late afternoon pattern’, respectively), genes involved in several specific cellular functions were highly enriched and predominant. These patterns of transcript abundance during a diel cycle provide insights for when some important metabolic processes might occur in ‘Ca. T. aerophilum’, and some specific examples will be described in the following discussion. However, because transcription obviously might not reflect translation, and translation might not reflect actual enzymatic activities, these patterns provide suggestions but do not allow one to reach definitive conclusions about the timing of metabolic activities in most cases.
Photosynthesis
The metagenome of ‘Ca. T. aerophilum’ encodes the photosynthetic reaction center proteins PscA, PsaB, PsaC, and PsaD; the BChl a-binding FMO protein (FmoA); and the BChl a-binding, chlorosome baseplate protein, CsmA. All of these proteins occur universally in GSB (Bryant et al., 2012). Transcript levels for these genes were up to 100-fold higher at night compared with daytime levels (Figure 4b, red line), which strongly suggests that the synthesis of reaction centers and chlorosomes mostly occurs at night. The most likely reason for this is the decrease in O2 levels beginning in late afternoon, when Synechococcus spp. switch from oxygenic photosynthesis to respiration and then fermentation (Nold and Ward, 1996, Steunou et al., 2006, Jensen et al., 2011, Figures 4a and ). Other genes in the ‘night pattern’ cluster included csmC, encoding another GSB-specific chlorosome envelope protein (Frigaard et al., 2004); pucC, whose product has various roles in photosynthesis (Tichy et al., 1991; Steunou et al., 2004; Jaschke et al., 2008); and genes neighboring csmA and csmC, whose probable functioning in photosynthesis is suggested by these and other data (Cao et al., 2012; Garcia Costas et al., 2012; Supplementary Figure 2).

Light and O2 conditions (a) and patterns of transcript levels for selected groups of genes of ‘Ca. T. aerophilum’ (b, c) in the Mushroom Spring microbial mat. Mean±s.e.m. were calculated and shown for each groups of genes. Genes of each group are listed in Supplementary Table 2. Patterns of transcript levels of a few Synechococcus genes (d) were also shown for comparison purposes and are not the focus of this study. Boxes indicate the early morning and late afternoon time periods discussed in the main text. Complex I, NADH dehydrogenase; ACIII, alternative complex III; Complex IV, cytochrome c oxidase.
Assuming that translation continues in ‘Ca. T. aerophilum’ cells during the night, it probably predominantly synthesizes and assembles reaction centers and chlorosomes at that time. However, phototrophic energy production is still likely to be highest during the day, simply because of the disparity in irradiance levels between these two periods (Figure 4). The possession of chlorosomes as light-harvesting antennae provides ‘Ca. T. aerophilum’ with the ability to harvest light efficiently at dawn and dusk, when irradiance levels are only a fraction of the maximal values (Figure 4a). Furthermore, it is worth noting that, at least during nights with clear skies, minimal fog from the source pool, and substantial moonlight, the potential exists for these organisms to continue some phototrophic energy production at night. The night-time downwelling quantum irradiance (400–700 nm) measured during the same expedition on 12 September 2009–13 September 2009 was up to 0.25 μmol photons m−2 s−1. For comparison, GSB living at the chemocline of the Black Sea experience in situ irradiances that are >100-fold lower: 0.00075–0.0022 μmol photons m−2 s−1. When tested under laboratory conditions, the carbon dioxide fixation rate for these organisms was saturated for light at an irradiance value of 1 μmol photons m−2 s−1 (Manske et al., 2005).
BChl biosynthesis
The ‘Ca. T. aerophilum’ metagenome contained all of the genes for BChl biosynthesis found in GSB except bchE, bchM and bchU. Suggesting its presence in ‘Ca. T. aerophilum’, transcripts for bchE were detected in the metatranscriptome (Table 1). Search results for bchM in the metatrancriptome were inconclusive (Table 1), but because every other Chl-producing organism possesses either bchM or chlM (Gomez Maqueo Chew and Bryant, 2007), chances are obviously high that this gene also occurs in ‘Ca. T. aerophilum’. No transcripts for a bchU gene similar in sequence to those of GSB were detected; however, bchU transcripts for other organisms producing BChl c (e.g. Chloroflexus spp. and ‘Ca. C. thermophilum’) were detected using the C. thalassium BchU sequence as the query (Table 1). Thus, ‘Ca. T. aerophilum’ probably does not have bchU, and therefore it should synthesize BChl d instead of BChl c as its major BChl for chlorosome assembly (Maresca et al., 2004). This deduction is very strongly supported by the detection of BChl d in pigment analyses of mat samples (Bauld and Brock, 1973; Pagel M, Ward DM, and Bryant DA, unpublished results). Furthermore, in situ transmitted light spectra show an absorption band at 730 nm characteristic of aggregated BChl d within the upper 1 mm of the mat (Ward et al., 2006, Kühl M, Jensen SI, Ward DM et al., unpublished results). Because the other two chlorosome-synthesizing organisms in the mat, ‘Ca. C. thermophilum’ and Chloroflexus spp., are known to synthesize BChl c (Bryant et al., 2007; Klatt et al., 2011; Bryant et al., 2012), ‘Ca. T. aerophilum’ is the most likely source of BChl d. Compared with chlorosomes containing BChl c, chlorosomes containing BChl d absorb light of shorter wavelength and are less effective at light harvesting at very low irradiance values (Maresca et al., 2004). This might explain why ‘Ca. T. aerophilum’ is found closer to the mat surface, whereas BChl c-synthesizing Chloroflexus spp. occur deeper in the mat (Ramsing et al., 2000, Klatt et al., 2011). This difference in the major antenna BChl should allow ‘Ca. T. aerophilum’ to occupy a different ecological niche than other chlorophototrophic populations, which synthesize Chl a, BChl c, or BChl a as major light-harvesting pigments (Klatt et al., 2011).
The ‘Ca. T. aerophilum’ metagenome also contained a gene, hemF, for heme and BChl biosynthesis, which has not previously been observed in any GSB genome. HemF is an O2-dependent coproporphyrinogen III oxidase (Xu and Elliott, 1993), and the presence of the hemF gene suggests that ‘Ca. T. aerophilum’ not only tolerates O2, but that it utilizes O2 as a substrate in one of its most important pathways. The presence of hemF strongly supports the conclusion that ‘Ca. T. aerophilum’ is an aerobe.
The patterns of transcript abundance for the bch and bci genes, which are responsible for the transformation of protoporphyrin IX to BChls, are shown in Figure 4b (green line). Transcripts for these genes were highest in the late afternoon and exhibited a secondary maximum in the early morning. These data suggested that more BChl biosynthesis enzymes are probably synthesized during these two light and O2 transition periods, which presumably reflects an increased demand for BChls at these (and later) times.
Central carbon metabolism
One of the most striking differences between ‘Ca. T. aerophilum’ and GSB was the absence of genes encoding ATP-dependent citrate lyase, which thus causes the reverse TCA cycle to be incomplete. The genes encoding the two subunits of this enzyme were not found in the metagenome, and even though these genes are extremely well conserved among GSB (Table 1), no transcripts were detected. Genes for alternative enzymes catalyzing this same reaction, namely citryl-CoA synthetase and citryl-CoA lyase (Aoshima et al., 2004a, 2004b), and the postulated type-II ATP citrate lyase (Hügler and Sievert, 2011), were also absent. Key enzymes of other CO2-fixing pathways such as Calvin-Benson-Bassham, 3-hydroxypropionate and Wood–Ljungdahl pathways were similarly not identified in the metagenome of ‘Ca. T. aerophilum’. Therefore, ‘Ca. T. aerophilum’ is unlikely to be an autotroph.
The ‘Ca. T. aerophilum’ metagenome includes genes required for the assimilation of acetate and propionate, which are principal fermentation products in the mat (Anderson et al., 1987; Nold and Ward, 1996). After activation of acetate by acetyl-CoA synthetase, acetyl-CoA could be carboxylated by pyruvate:ferredoxin oxidoreductase (NifJ) to form pyruvate, or it could enter the oxidative TCA cycle. After similar activation of propionate, propionyl-CoA could be assimilated through the 2-methylcitrate cycle, which converts propionyl-CoA to pyruvate (Horswill-Escalante-Semerena 1999), or it could be transformed into succinyl-CoA. ‘Ca. T. aerophilum’ encodes complete sets of genes for glycolysis and gluconeogenesis. It also encodes glycogen synthase and phosphorylase, which suggests that ‘Ca. T. aerophilum’ produces glycogen for carbon storage.
Two major transcript patterns were observed for the genes associated with core carbon metabolism, and the results are summarized in Figure 5. Transcripts for all genes associated with propionate assimilation were most abundant during the day, which suggested that ‘Ca. T. aerophilum’ likely photoassimilates propionate during the day. Transcripts of some other genes belonging to these core carbon metabolism pathways also exhibited increased transcript levels in the afternoon (i.e., an ‘afternoon pattern’). Transcripts for glycogen phosphorylase, and two glycolysis enzymes, increased in abundance in late afternoon, which suggested that glycogen is likely converted to phosphoenolpyruvate (PEP) at that time. Transcripts for several genes, whose products catalyze the transformation of acetate into oxaloacetate, also increased in the late afternoon. These trends strongly implied that an increase in carbon flux from assimilated acetate towards oxaloacetate occurs at that time during the diel cycle. The increased transcript abundance for several TCA cycle genes implied that oxaloacetate is probably transformed into other compounds in the TCA cycle. Because most of the enzymes in the cycle catalyze reversible reactions, it is difficult to pinpoint the end product from transcription data alone. However, it seems reasonable to hypothesize that 2-oxoglutarate is the major end product, because in the C-5 pathway it is the precursor metabolite required for BChl biosynthesis, which represents the major carbon sink in GSB (up to 30% of cell carbon in Chlorobaculum tepidum) (Montaño et al., 2003; Frigaard and Bryant, 2006; Gomez Maqueo Chew and Bryant, 2007; Saga et al., 2007; Liu and Bryant, 2012). Consistent with this hypothesis, transcript levels for genes encoding enzymes of BChl biosynthesis increased at the same time (Figure 4b, blue and green lines).

In situ patterns of transcript levels of genes involved in core carbon metabolism of ‘Ca. T. aerophilum’. Transcript levels for genes marked by red arrows increased during late afternoon (average pattern shown in Figure 4b, blue line). Those marked by blue arrows showed highest transcript levels during the day (‘day pattern’ in Figure 3). Those marked by black indicate unidentified genes. Most of the reactions are reversible but are thought to favor the directions shown in this figure (see text for details). acs, acetyl-CoA synthetase; bch, bci, BChl biosynthesis genes; can, carbonic anhydrase; dhnA, fructose-1,6-bisphosphate aldolase; gapA, glyceraldehyde-3-phosphate dehydrogenase; glgP, glycogen phosphorylase; gltA, citrate synthase; icd, isocitrate dehydrogenase, korAB, 2-oxoglutarate ferredoxin oxidoreductase; nifJ, pyruvate ferredoxin oxidoreductase; pck, PEP carboxykinase; ppsA, pyruvate phosphate dikinase; sdh, succinate dehydrogenase.
Oxaloacetate can be converted to 2-oxoglutarate by either or both of the two routes: the incomplete reductive TCA cycle, which is supported by increased transcript levels for succinate dehydrogenase and 2-oxoglutarate:ferredoxin oxidoreducatase genes; or the oxidative branch of the TCA cycle, which is supported by increased transcript levels for citrate synthase and isocitrate dehydrogenase genes. Simultaneous carbon flux through both the oxidative and reductive TCA branches catalyzed by the same enzymes discussed here has been demonstrated in C. tepidum during mixotrophic growth with acetate (Tang and Blankenship, 2010; Tang et al., 2011). The conversion of acetate into 2-oxoglutarate includes several anaplerotic CO2 fixation reactions, which requires an ample supply of CO2. Suggesting an increased demand for CO2 in the late afternoon, transcript levels for carbonic anhydrase increased at that time. The overall increase in transcript levels for some genes of central carbon metabolism in ‘Ca. T. aerophilum’ implies that increased carbon flux towards 2-oxoglutarate occurs in this organism during late afternoon. It should be noted that transcripts for citrate synthase remained relatively high from late afternoon into the night. This observation suggested that oxidation of acetyl-CoA by the TCA cycle likely extends into the night.
Electron transport complexes
The electron transport chains of ‘Ca. T. aerophilum’ and ‘Ca. C. thermophilum’ are strikingly similar (Garcia Costas et al., 2012) and both include type-1 NADH dehydrogenase (complex I), two cytochrome b-Rieske complexes (PetAB), alternative complex III (ACIII), and caa3-type cytochrome c oxidase (complex IV). Furthermore, transcript abundance patterns for the genes encoding the subunits of these complexes in ‘Ca. T. aerophilum’ were almost identical to those of ‘Ca. C. thermophilum’ (the results for ‘Ca. C. thermophilum’ are not shown here). These observations clearly imply that these organisms have similar physiology. Because ‘Ca. C. thermophilum’ is an aerobe (Bryant et al., 2007), ‘Ca. T. aerophilum’ is also likely to be an aerobe.
As in ‘Ca. C. thermophilum’, two copies of most of the type-1 NADH dehydrogenase genes were found in the metagenome. Both copies of these genes are closely related to those of other GSB. Whether the two sets of genes have different functions is unclear, but the transcript patterns were similar for both sets of genes. The average increase in transcript levels (>20-fold higher than night levels) for these genes was among the largest increases observed during the day (Figure 4c, red line).
One of the two cytochrome b-Rieske complexes, PetAI-PetBI, was similar to those of GSB; in these organisms this complex is thought to function as a menaquinol:cytochrome c oxidoreductase (Bryant et al., 2012). The other complex was encoded by petAII-petBII and an immediate downstream gene related to cydA, all of which had similar transcription patterns. Compared with the CydA subunit of cytochrome bd quinol oxidases, this CydA-like protein had an extra, C-terminal, monoheme cytochrome c-like domain. PetA and PetB proteins of both ‘Ca. T. aerophilum’ complexes and PetAIIBII-CydA of ‘Ca. C. thermophilum’ were most similar to homologs in GSB (Supplementary Figure 3a). The CydA-like protein was also similar to related proteins in ‘Ca. C. thermophilum’ and Deltaproteobacteria (Supplementary Figure 3b). It seems possible that a lateral gene transfer of the petAII, petBII and cydA-like genes may have occurred between ‘Ca. T. aerophilum’ and ‘Ca. C. thermophilum’. The function of the putative PetAII-PetBII-CydA complex is unknown, but the different transcription patterns for the petAII-petBII-cydA andpetAI-petBI operons strongly suggest that these two complexes have different roles in electron transport. Transcript levels for the petAI-petBI genes were among those showing the highest increase during daytime and exhibited changes of >100-fold compared with transcript levels at night. Transcript levels for the petAII-petBII-cydA genes were highest during the late afternoon (Figure 4c, orange and green lines).
As in ‘Ca. C. thermophilum’ (Garcia Costas et al., 2012), the genes encoding ACIII and caa3-type cytochrome c oxidase are clustered on the chromosome, and this suggests that ACIII functions in respiration. ACIII has been shown to be functionally identical to the cytochrome bc1 complex (that is, it acts as a quinol:cytochrome c oxidoreductase), and its coupling with cytochrome c oxidase has also been reported (Yanyushin et al., 2005; Gao et al., 2009). The transcript levels for the genes of these two complexes were similar and were highest in the afternoon (Figure 4c, blue lines), which suggests that increased respiration may be required at that time. Similar to observations made in Synechococcus spp. (Steunou et al., 2006, 2008), increased respiration could quickly decrease O2 concentration and protect oxygen-sensitive processes that require light energy.
Nitrogen metabolism
The ‘Ca. T. aerophilum’ metagenome did not include any nif genes, encoding MoFe-nitrogenase and its assembly proteins, which are highly conserved and universally present in GSB. No transcripts for the nifHDK genes derived from Chlorobiales were detected, even though the nif genes derived from other mat populations were readily detected using C. thalassium nitrogenase sequences as queries (Table 1). Because MoFe–nitrogenase and accessory protein sequences of GSB are very highly conserved, it is therefore highly unlikely that ‘Ca. T. aerophilum’ has nif genes. Consistent with this suggestion, the ABC transporter for molybdate, which is found in all GSB, was also missing in the metagenome of ‘Ca. T. aerophilum’ (Supplementary Table 1). The metagenome lacked assimilatory nitrate and nitrite reductases but included an ammonium transporter. These data suggested that ‘Ca. T. aerophilum’ obtains nitrogen either from the hot-spring water, which contains ∼4 μM ammonium (Papke et al., 2003, Holloway et al., 2011), or from one or more organisms in the mat community, or both. Although Roseiflexus spp. have genes related to nitrogenase and may potentially fix N2, Synechococcus spp. are currently the only confirmed N2-fixing chlorophototrophic organisms in the mat (Steunou et al., 2006, 2008).
Other pathways of interest
Although all GSB have the ability to oxidize sulfide, the enzymes involved in oxidative sulfur metabolism and the genes that encode these mostly multi-subunit enzymes are quite diverse (Gregersen et al., 2011). However, none of the genes encoding more than a dozen different enzymes for sulfur oxidation, which are almost universally found in GSB, were present in the metagenome of ‘Ca. T. aerophilum’ (Table 1). Furthermore, a search for transcripts of some of the most highly conserved genes also failed to identify any genes for sulfide oxidation. These results strongly suggested that ‘Ca. T. aerophilum’ cannot oxidize sulfide or other reduced sulfur species. Nevertheless, Octopus and Mushroom Springs do have low sulfate levels (∼200 μM; Papke et al., 2003, Holloway et al., 2011) that support sulfide production by sulfate-reducing bacteria, which have been detected in these mats (Dillon et al., 2007). Furthermore, ‘Ca. T. aerophilum’ lives at a depth, that is, the upper 1 mm of the mat, where O2 production by Synechococcus spp. leads to strong daytime supersaturation (Ramsing et al., 2000; Jensen et al., 2011; Figure 4a). Thus, except in low-light periods, sulfide concentrations are very low in the mat environment in the layers of the mat where ‘Ca. T. aerophilum’ lives. The ‘Ca. T. aerophilum’ metagenome also appeared to be missing genes for the use of H2, CO, Fe2+, nitrite, or arsenite as electron donors for CO2 fixation. These data provide further evidence that ‘Ca. T. aerophilum’ is a photoheterotroph, although anaplerotic CO2 fixation probably occurs through the incomplete reverse TCA cycle pathway as discussed above.
‘Ca. T. aerophilum’ lacked the genes required for the biosynthesis of several essential nutrients, including valine, leucine, isoleucine, possibly some other amino acids and vitamin B12 (Table 1, see Supplementary Information for a more detailed discussion). Interestingly, these genes were similarly missing in the phylogenetically distant acidobacterium, ‘Ca. C. thermophilum’ (Garcia Costas et al., 2012). We speculate that these molecules or their precursors are synthesized by other organisms in the mat and are available for uptake and assimilation. These observations provide additional strong support for the conclusion that ‘Ca. T. aerophilum’ is a photoheterotroph.
Cellular overview and potential interactions with other organisms
Figure 6 summarizes the dramatically different physiological properties distinguishing Ca. T. aerophilum from previously characterized GSB—foremost that it is almost certainly an aerobic photoheterotroph. The overall similarity of electron transport complexes in ‘Ca. T. aerophilum’ to those of the aerobic acidobacterium, ‘Ca. C. thermophilum’, especially the presence of complexes associated with aerobic respiration and the presence of O2-utilizing HemF, strongly suggest that ‘Ca. T. aerophilum’ also is an aerobe. To emphasize this important distinction, we suggest that its species epithet be ‘aerophilum’. Moreover, because it has an incomplete reductive TCA cycle and cannot oxidize common inorganic electron donors including sulfide, ‘Ca. T. aerophilum’ lacks the ability to fix CO2 autotrophically. It also lacks the ability to fix N2. Thus, ‘Ca. T. aerophilum’ must depend on other organisms in the mat for some essential nutrients, including amino acids, vitamin B12, and possibly reduced nitrogen. Although further evidence to confirm these tentative conclusions concerning the physiology of ‘Ca. T. aerophilum’ are necessary from cultivation and biochemical studies, these deductions will greatly aid attempts to isolate this organism. Without this information and only based on 16S rRNA data, one would probably select the standard, anoxic enrichment conditions with sulfide used for the cultivation of GSB, yet these conditions would be highly unfavorable for the growth of ‘Ca. T. aerophilum’. The physiological and metabolic insights gained for this phylogenetically distinctive but yet uncultured population are an excellent example of how culture-independent methods, employing high-throughput sequencing approaches, can quickly change our perception of a group of seemingly well-defined bacteria. This study also underscores the importance of continuing efforts to determine the properties of organisms detected by 16S rRNA sequences but belonging to divergent groups in unconventional environments.

Cellular overview of the putative metabolic capability of ‘Ca. T. aerophilum’ emphasizing metabolisms common in GSB, but not present in ‘Ca. T. aerophilum’. Only pathways, enzymes or transporters of interest are shown here. Enzymes/pathways that are conserved in GSB but missing in ‘Ca. T. aerophilum’ are marked by red X’s. Blue arrows mark the flow of electrons.
Supplementary Figure 4 summarizes a conceptual model summarizing when and how interactions between ‘Ca. T. aerophilum’ and other organisms in the mat might occur according to the metatranscriptomic data. It is most likely that the synthesis of chlorosomes and reaction centers occurs during night, because the very high levels of transcripts for genes encoding reaction centers, FMO, and chlorosomes have no value unless they are converted into protein products. Other than this, it is difficult to predict what ‘Ca. T. aerophilum’ is doing metabolically at night from the transcription data. Fermentation products released by Synechococcus spp. could be taken up and respired (provided that a small amount of O2 (below detection limit) penetrates into the upper few 100 μm of the mat). We also hypothesize that moonlight, when available, could contribute some energy for maintenance metabolism on clear nights (Supplementary Figure 4a).
Assembly of the photosynthetic apparatus at night would require a substantial supply of BChls to exist at this time. These molecules are probably synthesized mainly during late afternoon, and secondarily in the early morning (Supplementary Figure 4b). The energy level at night might not be sufficient to support the synthesis of numerous antenna BChl molecules, and O2-sensitive BChls might be degraded during the day unless they are assembled into complexes. In comparison, during late afternoon and early morning, the solar irradiance is still sufficiently high to saturate phototrophic energy production in a chlorosome-containing organism such as ‘Ca. T. aerophilum’ while O2 concentrations are moderate (Figure 4a). Furthermore, new carbon and nitrogen sources from Synechococcus fermentation and N2 fixation are also available during those times (Figure 4d; Steunou et al., 2006, 2008). Transcription patterns of genes involved in BChl synthesis and related carbon metabolism support this possibility.
Transcript levels for most genes and total mRNA production were highest for ‘Ca. T. aerophilum’ during the day. The synthesis of most cellular materials presumably also occurs at the same time, likely using photoassimilated acetate and propionate as the carbon source. Glycolate from photorespiration by Synechococcus spp. (Bateson and Ward, 1988) is also a possible carbon and electron source (see Supplementary Information). Transcript levels for glycogen synthase were also highest during the day; this suggests that sufficient carbon is available during the day to support its storage as glycogen (Supplementary Figure 4c).
The physiological inferences and the metabolic model derived from the metagenome and metatranscriptomic data (Figure 6 and Supplementary Figure 4) will require confirmation from a variety of additional experimental approaches, including proteomics, metabolomics, enzyme activity assays, and in situ stable-isotope labeling. The information gained in the present analyses will also enhance efforts to cultivate ‘Ca. T. aerophilum’ in the laboratory. The model presented here provides an initial framework for testing the complex metabolic and physiological dynamics that occur in this hot-spring microbial mat community. This study serves as an excellent example of the potential inherent in the metatranscriptomic method for validating as well as inferring the functions of uncultured members of complex communities.
References
Anderson KL, Tayne TA, Ward DM . (1987). Formation and fate of fermentation products in hot spring cyanobacterial mats. Appl Environ Microbiol 53: 2343–2352.
Aoshima M, Ishii M, Igarashi Y . (2004a). A novel enzyme, citryl-CoA synthetase, catalysing the first step of the citrate cleavage reaction in Hydrogenobacter thermophilus TK-6. Mol Microbiol 52: 751–761.
Aoshima M, Ishii M, Igarashi Y . (2004b). A novel enzyme, cirtyl-CoA lyase, catalysing the second step of the citrate cleavage reaction in Hydrogenobacter thermophilus TK-6. Mol Microbiol 52: 763–770.
Bateson MM, Ward DM . (1988). Photoexcretion and fate of glycolate in a hot spring cyanobacterial mat. Appl Environ Microbiol 54: 1738–1743.
Bauld J, Brock TD . (1973). Ecological studies of Chloroflexis, a gliding photosynthetic bacterium. Arch Microbiol 92: 267–284.
Bhaya D, Grossman AR, Steunou AS, Khuri N, Cohan FM, Hamamura N et al. (2007). Population level functional diversity in a microbial community revealed by comparative genomics and metagenomic analyses. ISME J 1: 703–713.
Bryant DA, Frigaard NU . (2006). Prokaryotic photosynthesis and phototrophy illuminated. Trends Microbiol 14: 488–496.
Bryant DA, Garcia Costas AM, Maresca JA, Gomez Maqueo Chew A, Klatt CG, Bateson MM et al. (2007). Candidatus Chloracidobacterium thermophilum: an aerobic phototrophic acidobacterium. Science 317: 523–526.
Bryant DA, Liu Z, Li T, Zhao F, Garcia Costas AM, Klatt CG et al. (2012). Comparative and functional genomics of anoxygenic green bacteria from the taxa Chlorobi, Chloroflexi, and Acidobacteria. In: RL Burnap, W Vermaas (eds). Advances in Photosynthesis and Respiration, Vol. 35, Functional Genomics and Evolution of Photosynthetic Systems. Springer, Dordrecht, The Netherlands, pp 47–102.
Buchanan BB, Arnon DI . (1990). A reverse KREBS cycle in photosynthesis: consensus at last. Photosynth Res 24: 47–53.
Cao L, Schepmoes AA, Vogl K, Bryant DA, Smith RD, Lipton MS et al. (2012). Comparison of Chloroflexus auranticaus strain J-10-fl proteomes of cells grown chemoheterotrophically and photoheterotrophically. Photosynth Res 110: 153–168.
Ciccarelli FD, Doerks T, von Mering C, Creevey CJ, Snel B, Bork P . (2006). Toward automatic reconstruction of a highly resolved tree of life. Science 311: 1283–1287.
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ et al. (2009). The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucl Acids Res 37: D141–D145.
Dillon JG, Fishbain S, Miller SR, Bebout BM, Habicht KS, Webb SM et al. (2007). High rates of sulfate reduction in a low-sulfate hot spring microbial mat are driven by a low level of diversity of sulfate-respiring microorganisms. Appl Environ Microbiol 73: 5218–5226.
Ferris MJ, Muyzer G, Ward DM . (1996). Denaturing gradient gel electrophoresis profiles of 16S rRNA-defined populations inhabiting a hot spring microbial mat community. Appl Envrion Microbiol 62: 340–346.
Ferris MJ, Ward DM . (1997). Seasonal distributions of dominant 16S rRNA-defined populations in a hot spring microbial mat examined by denaturing gradient gel electrophoresis. Appl Environ Microbiol 63: 1375–1381.
Fouke BW . (2011). Hot-spring systems geobiology: abiotic and biotic influences on travertine formation at Mammoth Hot Springs, Yellowstone National Park, USA. Sedimentology 58: 170–219.
Fouke BW, Bonheyo GT, Sanzenbacher E, Frias-Lopez J . (2003). Partitioning of bacterial communities between travertine depositional facies at Mammoth Hot Springs, Yellowstone National Park, USA. Can J Earth Sci 40: 1531–1548.
Frigaard NU, Bryant DA . (2006). Chlorosomes: antenna organelles in photosynthetic green bacteria. In: JM Shively (ed). Complex Intracellular Structures in Prokaryotes Vol. 2. Springer-Verlag, Berlin Heidelberg, pp 80–114.
Frigaard NU, Dahl C . (2009). Sulfur metabolism in phototrophic sulfur bacteria. Adv Microb Physiol 54: 103–200.
Frigaard NU, Li H, Milks KJ, Bryant DA . (2004). Nine mutants of Chlorobium tepidum each unable to synthesize a different chlorosome protein still assemble functional chlorosomes. J Bacteriol 186: 646–653.
Gao X, Xin Y, Blankenship RE . (2009). Enzymatic activity of the alternative complex III as a menaquinol:auracyanin oxidoreductase in the electron transfer chain of Chloroflexus aurantiacus. FEBS Lett 583: 3275–3279.
Garcia Costas AM, Liu Z, Tomsho LP, Schuster SC, Ward DM, Bryant DA . (2012). Complete genome of ‘Candidatus Chloracidobacterium thermophilum,’ a chlorophyll-based photoheterotroph from the phylum Acidobacteria. Environ Microbiol 14: 177–190.
Gomez Maqueo Chew A, Bryant DA . (2007). Chlorophyll biosynthesis in bacteria: the origins of structural and functional diversity. Annu Rev Microbiol 61: 113–129.
Gregersen LH, Bryant DA, Frigaard NU . (2011). Mechanisms and evolution of oxidative sulfur metabolism in green sulfur bacteria. Front Microbiol 2: 116.
Heising S, Richter L, Ludwig W, Schink B . (1999). Chlorobium ferrooxidans sp. nov., a phototrophic green sulfur bacterium that oxidizes ferrous iron in coculture with a ‘Geospirillum’ sp. strain. Arch Microbiol 172: 116–124.
Holloway JM, Nordstrom DK, Böhlke JK, McCleskey RB, Ball JW . (2011). Ammonium in thermal waters of Yellowstone National Park: processes affecting speciation and isotope fractionation. Geochim Cosmochim Acta 75: 4611–4636.
Horswill AR, Escalante-Semerena JC . (1999). Salmonella typhimurium LT2 catabolizes propionate via the 2-methylcitric acid cycle. J Bacteriol 181: 5615–5623.
Hügler M, Sievert SM . (2011). Beyond the Calvin cycle: autotrophic carbon fixation in the ocean. Ann Rev Mar Sci 3: 261–289.
Iino T, Mori K, Uchino Y, Nakagawa T, Harayama S, Suzuki K . (2010). Ignavibacterium album gen. nov., sp. nov., a moderately thermophilic anaerobic bacterium isolated from microbial mats at a terrestrial hot spring and proposal of Ignavibacteria classis nov., for a novel lineage at the periphery of green sulfur bacteria. Int J Syst Evol Microbiol 60: 1376–1382.
Imhoff JF . (2003). Phylogenetic taxonomy of the family Chlorobiaceae on the basis of 16S rRNA and fmo (Fenna-Matthews-Olson protein) gene sequences. Int J Syst Evol Microbiol 53: 941–951.
Imhoff JF, Thiel V . (2010). Phylogeny and taxonomy of Chlorobiaceae. Photosynth Res 104: 123–136.
Jaschke PR, Leblanc HN, Lang AS, Beatty JT . (2008). The PucC protein of Rhodobacter capsulatus mitigates an inhibitory effect of light-harvesting 2 alpha and beta proteins on light-harvesting complex 1. Photosynth Res 95: 279–284.
Jensen SI, Steunou AS, Bhaya D, Kühl M, Grossman AR . (2011). In situ dynamics of O2, pH and cyanobacterial transcripts associated with CCM, photosynthesis and detoxification of ROS. ISME J 5: 317–328.
Klatt CG, Wood JM, Rusch DB, Bateson MM, Heidelberg JF, Bhaya D et al. (2011). Community ecology of hot spring cyanobacterial mats: predominant populations and their functional potential. ISME J 5: 1262–1278.
Lau MC, Aitchison JC, Pointing SB . (2009). Bacterial community composition in thermophilic microbial mats from five hot springs in central Tibet. Extremophiles 13: 139–149.
Li H, Jubilerer S, Garcia Costas AM, Frigaard N-U, Bryant DA . (2009). Multiple antioxidant proteins protect Chlorobaculum tepidum against oxygen and reactive oxygen species. Arch Microbiol 91: 853–867.
Liu Z, Bryant DA . (2012). Bacteriochlorophyll biosynthesis and assembly in green chlorophototrophic bacteria: theme and variations. In: KM Kadish, KM Smith, R Guilard (eds). Handbook of Porphyrin Science. World Scientific Publishers, Hackensack, NJ, USA, (in press).
Liu Z, Klatt CG, Wood JM, Rusch DB, Ludwig M, Wittekindt N et al. (2011). Metatranscriptomic analyses of chlorophototrophs of a hot-spring microbial mat. ISMEJ 5: 1279–1290.
Ludwig W, Klenk HP . (2001). Overview: a phylogenetic backbone and taxonomic framework for prokaryotic systematics. In: DR Boone, RW Castenholz (eds) Bergey’s Manual of Systematic Bacteriology 2nd edn Vol. 1. Springer, Berlin, pp 49–65.
Manske AK, Glaeser J, Kuypers MM, Overmann J . (2005). Physiology and phylogeny of green sulfur bacteria forming a monospecific phototrophic assemblage at a depth of 100 meters in the Black Sea. Appl Environ Microbiol 71: 8049–8060.
Maresca JA, Gomez Maqueo Chew A, Ponsatí MR, Frigaard NU, Ormerod JG, Bryant DA . (2004). The bchU gene of Chlorobium tepidum encodes the C-20 methyltransferase in bacteriochlorophyll c biosynthesis. J Bacteriol 186: 2558–2566.
Montaño GA, Bowen BP, LaBelle JT, Woodbury NW, Pizziconi VB, Blankenship RE . (2003). Characterization of Chlorobium tepidum chlorosomes: a calculation of bacteriochlorophyll c per chlorosome and oligomer modeling. Biophys J 85: 2560–2565.
Nold SC, Ward DM . (1996). Photosynthate partitioning and fermentation in hot spring microbial mat communities. Appl Environ Microbiol 62: 4598–4607.
Overmann J . (2001). Green sulfur bacteria. In: DR Boone, RW Castenholz (eds). Bergey’s Manual of Systematic Bacteriology 2nd edn Vol 1. Springer, Berlin, pp 601–605.
Overmann J . (2008). Ecology of phototrophic sulfur bacteria. In: R Hell, C Dahl, DB Knaff, T Leustek (eds) Sulfur Metabolism in Phototrophic Organisms. Springer, Dordrecht, The Netherlands, pp 375–396.
Papke RT, Ramsing NB, Bateson MM, Ward DM . (2003). Geographical isolation in hot spring cyanobacteria. Environ Microbiol 5: 650–659.
Ramsing NB, Ferris MJ, Ward DM . (2000). Highly ordered vertical structure of Synechococcus populations within the one-millimeter-thick photic zone of a hot spring cyanobacterial mat. Appl Environ Microbiol 66: 1038–1049.
Saga Y, Shibata Y, Itoh S, Tamiaki H . (2007). Direct counting of submicrometer-sized photosynthetic apparatus dispersed in medium at cryogenic temperature by confocal laser fluorescence microscopy: estimation of the number of bacteriochlorophyll c in single light-harvesting antenna complexes chlorosomes of green photosynthetic bacteria. J Phys Chem B 111: 12605–12609.
Steunou AS, Bhaya D, Bateson MM, Melendrez MC, Ward DM, Brecht E et al. (2006). In situ analysis of nitrogen fixation and metabolic switching in unicellular thermophilic cyanobacteria inhabiting hot spring microbial mats. Proc Natl Acad Sci USA 103: 2398–2403.
Steunou AS, Jensen SI, Brecht E, Becraft ED, Bateson MM, Kilian O et al. (2008). Regulation of nif gene expression and the energetics of N2 fixation over the diel cycle in a hot spring microbial mat. ISME J 2: 364–378.
Steunou AS, Ouchane S, Reiss-Husson F, Astier C . (2004). Involvement of the C-terminal extension of the alpha polypeptide and of the PucC protein in LH2 complex biosynthesis in Rubrivivax gelatinosus. J Bacteriol 186: 3143–3152.
Tamura K, Dudley J, Nei M, Kumar S . (2007). MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24: 1596–1599.
Tang KH, Blankenship RE . (2010). Both forward and reverse TCA cycles operate in green sulfur bacteria. J Biol Chem 285: 35848–35854.
Tang KH, Tang YJ, Blankenship RE . (2011). Carbon metabolic pathways in phototrophic bacteria and their broader evolutionary implications. Front Microbiol 2: 165.
Teeling H, Meyerdierks A, Bauer M, Amann R, Glöckner FO . (2004). Application of tetranucleotide frequencies for the assignment of genomic fragments. Environ Microbiol 6: 938–947.
Tichy HV, Albien KU, Gad’on N, Drews G . (1991). Analysis of the Rhodobacter capsulatus puc operon: the pucC gene plays a central role in the regulation of LHII (B800-850 complex) expression. EMBO J 10: 2949–2955.
Wahlund TM, Madigan MT . (1993). Nitrogen fixation by the thermophilic green sulfur bacterium Chlorobium tepidum. J Bacteriol 175: 474–478.
Wahlund TM, Tabita FR . (1997). The reductive tricarboxylic acid cycle of carbon dioxide assimilation: initial studies and purification of ATP-citrate lyase from the green sulfur bacterium Chlorobium tepidum. J Bacteriol 179: 4859–4867.
Ward DM, Bateson MM, Ferris MJ, Kühl M, Wieland A, Koeppel A et al. (2006). Cyanobacterial ecotypes in the microbial mat community of Mushroom Spring (Yellowstone National Park, Wyoming) as species-like units linking microbial community composition, structure and function. Phil Trans R Soc B 361: 1997–2008.
Ward DM, Ferris MJ, Nold SC, Bateson MM . (1998). A natural view of microbial biodiversity within hot spring cyanobacterial mat communities. Microbiol Mol Biol Rev 62: 1353–1370.
Weller R, Bateson MM, Heimbuch BK, Kopczynski ED, Ward DM . (1992). Uncultivated cyanobacteria, Chloroflexus-like inhabitants, and spirochete-like inhabitants of a hot spring microbial mat. Appl Environ Microbiol 58: 3964–3969.
Xu K, Elliott T . (1993). An oxygen-dependent coproporphyrinogen oxidase encoded by the hemF gene of Salmonella typhimurium. J Bacteriol 175: 4990–4999.
Yanyushin MF, del Rosario MC, Brune DC, Blankenship RE . (2005). New class of bacterial membrane oxidoreductases. Biochemistry 44: 10037–10045.
Acknowledgements
DAB and DMW acknowledge support from the NASA Exobiology Program (NX09AM87G) and from the Genomic Science Program (GSP) of the US Department of Energy (DOE), Office of Biological and Environmental Research (BER). This contribution originates from the GSP Foundational Scientific Focus Area (FSFA) of the Pacific Northwest National Laboratory. DAB also acknowledges support from the National Science Foundation (MCB-0523100), Department of Energy (DE-FG02-94ER20137), and the DOE-Joint Genome Institute. MK acknowledges support form the Danish Natural Science Research Council. DMW also acknowledges support from the National Science Foundation Frontiers in Integrative Biology Research Program (EF-0328698) and the IGERT Program in Geobiological Systems (DGE 0654336). This study was conducted under Yellowstone National Park research permits YELL-0129 and 5494 (DMW) and YELL-02058 (MK), and we greatly appreciate the assistance of National Park Service personnel. The work conducted by the DOE-Joint Genome Institute is supported by the Office of Science of the US Department of Energy under Contract No. DE-AC02-05CH11231. We appreciate the assistance of Eric D Becraft, Shane Nowack, Jason Wood and Laurey Steinke during the field expedition.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Rights and permissions
About this article
Cite this article
Liu, Z., Klatt, C., Ludwig, M. et al. ‘Candidatus Thermochlorobacter aerophilum:’ an aerobic chlorophotoheterotrophic member of the phylum Chlorobi defined by metagenomics and metatranscriptomics. ISME J 6, 1869–1882 (2012). https://doi.org/10.1038/ismej.2012.24
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2012.24
Keywords
This article is cited by
-
Late acquisition of the rTCA carbon fixation pathway by Chlorobi
Nature Ecology & Evolution (2023)
-
Acclimation of the photosynthetic apparatus to low light in a thermophilic Synechococcus sp. strain
Photosynthesis Research (2022)
-
Remarkably coherent population structure for a dominant Antarctic Chlorobium species
Microbiome (2021)
-
Reaction centers of the thermophilic microaerophile, Chloracidobacterium thermophilum (Acidobacteria) I: biochemical and biophysical characterization
Photosynthesis Research (2019)
-
15N photo-CIDNP MAS NMR analysis of reaction centers of Chloracidobacterium thermophilum
Photosynthesis Research (2018)
