
Abstract
Ticks transmit a variety of viral, bacterial and protozoal pathogens, which are often zoonotic. The aim of this study was to identify diverse tick microbiomes, which may contain as-yet unidentified pathogens, using a metagenomic approach. DNA prepared from bacteria/archaea-enriched fractions obtained from seven tick species, namely Amblyomma testudinarium, Amblyomma variegatum, Haemaphysalis formosensis, Haemaphysalis longicornis, Ixodes ovatus, Ixodes persulcatus and Ixodes ricinus, was subjected to pyrosequencing after whole-genome amplification. The resulting sequence reads were phylotyped using a Batch Learning Self-Organizing Map (BLSOM) program, which allowed phylogenetic estimation based on similarity of oligonucleotide frequencies, and functional annotation by BLASTX similarity searches. In addition to bacteria previously associated with human/animal diseases, such as Anaplasma, Bartonella, Borrelia, Ehrlichia, Francisella and Rickettsia, BLSOM analysis detected microorganisms belonging to the phylum Chlamydiae in some tick species. This was confirmed by pan-Chlamydia PCR and sequencing analysis. Gene sequences associated with bacterial pathogenesis were also identified, some of which were suspected to originate from horizontal gene transfer. These efforts to construct a database of tick microbes may lead to the ability to predict emerging tick-borne diseases. Furthermore, a comprehensive understanding of tick microbiomes will be useful for understanding tick biology, including vector competency and interactions with pathogens and symbionts.
Similar content being viewed by others

Introduction
Ticks (Acari: Ixodida) are amongst the most well-known blood-sucking arthropods. They can transmit a variety of pathogens, including viruses, bacteria and protozoa, to humans and animals. Opportunities for human/animal contact in natural foci of tick-borne infection are increasing, along with expanded distributions of vector ticks due to environmental changes (Parola, 2004; Beugnet and Marié, 2009). Meanwhile, reports of novel tick-borne pathogens are also increasing (Spolidorio et al., 2010; Yu et al., 2011). Above all, many of the emerging rickettsial pathogens identified in recent decades were first discovered in ticks and subsequently recognized as the causative agents of animal or human diseases (Parola et al., 2005). This indicates that active surveillance for potential pathogens in ticks may be a feasible approach to predict and combat emerging tick-borne diseases. Given the diversity of tick species and their wide geographic distribution (Bowman and Nuttall, 2008), it is likely that as-yet unidentified pathogens are harboured by ticks.
Morphological detection by electron microscopy has shown that Rickettsia- and Wolbachia-like bacteria infect a variety of tick species (Roshdy, 1968; Lewis, 1979), although these bacteria are not necessarily pathogenic in either mammalian hosts or ticks. The availability of molecular genetic tools such as PCR and real-time PCR has increased the number of reports on a variety of microorganisms in ticks (Noda et al., 1997; Benson et al., 2004; van Overbeek et al., 2008); however, their biological roles and potential to cause disease in mammals remain poorly understood. Interactions between arthropods and their symbionts have been relatively well studied in aphids. A variety of symbionts are reported to have detrimental, neutral or beneficial effects on the aphid hosts; for example, through pathogenicity (Grenier et al., 2006), providing nutrition (Nakabachi and Ishikawa, 1999), altering gene expression associated with body colour (Tsuchida et al., 2010) and, most intriguingly, displaying protective functions against pathogens and predators (Scarborough et al., 2005). It has been suggested that certain symbiotic microbes might also affect the vector competency of their arthropod host (Weiss and Aksoy, 2011). A full characterization of microorganisms harboured by ticks will provide valuable information towards a better understanding of tick biology in terms of the relationship between ticks and their microbiomes, including microbes that are potential pathogens in mammals.
Recent progress in second generation sequencing technologies has led to the discovery of previously unknown organisms in a variety of samples, including arthropods (Cox-Foster et al., 2007; Warnecke et al., 2007; Suen et al., 2010), since uncultured microorganisms represent the vast majority of symbiotic, commensal and pathogenic microorganisms. Attempts to assess the microbial diversity in two tick species, Rhipicephalus microplus and Ixodes ricinus, using novel sequencing technologies, resulted in the identification of microbes (including bacterial species) previously unreported in ticks (Andreotti et al., 2011; Carpi et al., 2011). However, since the data processing methods used in these two studies were based on conventional homology-based sequence searches, the discoveries were limited to microorganisms for which genomic information was already available in the database used. An alternative approach to characterizing DNA fragments might offer improvements for investigating microbial diversity in ticks that are expected to harbour poorly characterized microorganisms.
Batch Learning Self-Organizing Map (BLSOM) is a bioinformatics method that follows a learning process and generates a map independently of the order of data input (Kanaya et al., 2001; Abe et al., 2003). This method was designed to separate and cluster genomic sequence fragments based on the similarity of oligonucleotide frequencies without any other taxonomical information. Since BLSOM does not require orthologous sequence sets and sequence alignments, it could provide a new systematic strategy for revealing the microbial diversity and relative abundance of different phylotype members of uncultured microorganisms in a wide variety of environmental metagenomic samples (Abe et al., 2005; Uehara et al., 2011). Of note, similar SOM-based methods were recently used to obtain clear phylotype-specific classification of metagenomic sequences (Chan et al., 2008; Martin et al., 2008; Dick et al., 2009; Weber et al., 2011).
In the present study, we used BLSOM to characterize diverse microbiomes in seven tick species. The ticks harboured a variety of bacteria, including those previously reported to be associated with human and animal diseases, and others not previously reported from ticks. This study was intended as a first step towards the discovery of emerging tick-borne pathogens and a systematic understanding of the relationship between ticks and their microbiomes.
Materials and methods
Tick species
Ticks were collected from the field by dragging flannel sheets over the vegetation. Three tick species, namely Amblyomma testudinarium (AT), Haemaphysalis formosensis (HF) and Haemaphysalis longicornis (HL) were collected in March 2009 in Miyazaki, Japan. This is an area where Rickettsia japonica, the causative agent of Japanese spotted fever, and Rickettsia tamurae, the spotted fever group rickettsia recently associated with human disease (Imaoka et al., 2011), are known to be endemic (Morita et al., 1990; S Yamamoto, personal communication). Two Ixodes species, Ixodes ovatus (IO) and Ixodes persulcatus (IP), were collected in June 2010 in Hokkaido, Japan, where Borrelia burgdorferi sensu lato (s.l.) and Anaplasma phagocytophilum, the causative agents of Lyme disease and human granulocytic anaplasmosis, respectively, are endemic (Miyamoto et al., 1992; Murase et al., 2011). Ixodes ricinus (IR) was collected in August 2010 in Soesterberg, The Netherlands, where Lyme disease is endemic (Hofhuis et al., 2006). Amblyomma variegatum (AV) ticks were collected in The Gambia in 2005 and subsequently maintained under laboratory conditions at the Utrecht Centre for Tick-borne Diseases, The Netherlands. Adult AV ticks from the second laboratory generation were used in this study. The collected ticks were pooled according to species, life stage and sex (except for HF and HL) and specific pool IDs (AVf, AVm, IOf, IOm, IPf, IPm, IRf, ATn, HFfmn and HLfmn) were provided as indicated in Table 1 (‘f’, ‘m’ and ‘n’ represent female, male and nymph, respectively).
DNA preparation
The live ticks were washed twice with 70% ethanol supplemented with 1% povidone-iodine (Meiji Seika, Tokyo, Japan) solution and rinsed three times with distilled water to decontaminate the tick body surface. Ticks were then ground with 4.8 mm stainless steel beads (TOMY, Tokyo, Japan) using the Micro Smash MS-100R (TOMY). Tick homogenates were treated with a membrane lysis buffer [10 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10 mM MgCl2, 0.1% IGEPAL CA-630 (Sigma Chemical Co., St. Louis, MO)] for 10 min on ice and centrifuged at 400 × g for 25 min. The supernatant was centrifuged at 20 000 × g for 30 min and the pellet containing the bacterial/archaeal cells was resuspended in a DNase buffer [25 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 0.8 U/μl DNase I (Takara, Shiga, Japan)] to remove any contaminating host-cell DNA. After 60 min at 37 °C, the reaction was stopped by adding 25 mM EDTA (pH 8.0). The solution was filtered through a 5.0 μm pore-size membrane filter (Millipore, Bedford, MA) by centrifugation at 1000 × g. The filtrate was centrifuged at 20 000 × g for 30 min and the resulting pellet was washed twice with PBS. All centrifugation steps were performed at 4 °C. To lyse a wide range of bacterial/archaeal species, this study utilized an achromopeptidase, which is reported to have broad-spectrum bacteriolytic activity (Ezaki and Suzuki, 1982). The bacterial/archaeal pellet was treated with an achromopeptidase buffer [10 mM Tris-HCl (pH 8.0), 10 mM NaCl, 1 U/μl achromopeptidase (WAKO, Osaka, Japan)] for 60 min at 37 °C. Bacterial/archaeal DNA was then extracted using the NucleoSpin Tissue XS Kit (Macherey-Nagel, Düren, Germany) according to the manufacturer′s instructions. The genomic DNA was then subjected to whole-genome amplification using the GenomiPhi V2 DNA Amplification Kit (GE Healthcare, Chalfont St Giles, UK) according to the manufacturer’s instructions. The DNA concentration was measured using the Quant-iT dsDNA BR assay with a Qubit Fluorometer (Invitrogen, Carlsbad, CA). All procedures were conducted under sterile conditions in a flow cabinet. A schematic flow diagram showing the extraction process of bacterial/archaeal DNA from ticks is presented in Figure 1.

Workflow for bacterial/archaeal purification and metagenomic analysis. The present strategy employed a process for purifying of bacteria/archaea, which comprised centrifugation, DNase treatment and filtration, to enrich bacterial/archaeal cells from tick homogenates prior to whole-genome amplification and pyrosequencing.
Pyrosequencing
Genomic DNA from each tick pool was subjected to pyrosequencing on a Roche/454 Genome Sequencer FLX Titanium (Roche Applied Science/454 Life Science, Brandford, CT) at Hokkaido System Science (Sapporo, Japan). Two GS FLX shotgun libraries were prepared using a standard protocol from the manufacturer and analysed in four independent sequencing runs: the first library contained three pools (ATn, HFfmn and HLfmn) with different multiplex identifiers and was analysed on 1/8 of the PicoTiterPlate (Roche Applied Science/454 Life Science), while the second library contained seven pools (AVf, AVm, IOf, IOm, IPf, IPm and IRf) and was analysed on 1/4 of the plate. The metagenomic sequences were deposited in the DNA Data Bank of Japan (DDBJ) (http://www.ddbj.nig.ac.jp) Sequence Read Archive under the accession no. DRA000590.
BLSOM analysis
Self-Organizing Map (SOM) is an unsupervised neural network algorithm that implements a characteristic nonlinear projection from the high-dimensional space of input data onto a two-dimensional array of weight vectors (Kohonen, 1990). We used the ‘Batch Learning SOM’ (BLSOM), which is a modified version of the conventional SOM for genome informatics that makes the learning process and creation of the resulting map independent of the order of data input (Kanaya et al., 2001; Abe et al., 2003). The initial weight vectors were defined by principal component analysis instead of random values. BLSOM learning was conducted as described previously (Abe et al., 2003), and the BLSOM program was obtained from UNTROD Inc., Japan (y_wada@nagahama-i-bio.ac.jp).
To estimate phylotypes of the metagenomic sequences, three types of large-scale BLSOMs, namely Kingdom-, Bacteria/Archaea- and Genus group-BLSOM, were constructed in advance, using sequences deposited in DDBJ/EMBL/GenBank as previously described (Abe et al., 2003). The clustering conditions were adopted from the previously optimised parameters to minimise the computation time without loss of clustering power (Abe et al., 2005). Kingdom-BLSOM was constructed with tetranucleotide frequencies for 5-kb sequences from the whole-genome sequences of 111 eukaryotes, 2813 bacteria/archaea, 1728 mitochondria, 110 chloroplasts and 31 486 viruses. To obtain more detailed phylotype information for bacterial/archaeal sequences, Bacteria/Archaea- and Genus group-BLSOM were constructed with a total of 3 500 000 5-kb sequences from 3157 species, for which at least 10 kb of sequence was available from DDBJ/EMBL/GenBank.
Mapping of metagenomic sequences longer than 300 bp on Kingdom-BLSOMs, after normalization of the sequence length, was conducted by finding the lattice point with the minimum Euclidean distance in the multidimensional space. To identify further detailed phylogenies of the metagenomic sequences that had been mapped to the bacterial/archaeal territories on Kingdom-BLSOM, these sequences were successively mapped on Bacteria/Archaea-BLSOM. Similar stepwise mappings of metagenomic sequences on the Genus group-BLSOM were subsequently conducted to obtain further detailed phylogenetic information. In order to evaluate the accuracy of BLSOMs for taxonomic classification of metagenomic sequences, the BLSOM analysis was applied to three previously published simulated datasets of varying complexities (Mavromatis et al., 2007). When sequences longer than 300 bp were mapped, approximately 90%, 70% and 40%–50% were correctly classified to the kingdom, phylum and genus levels, respectively (Supplementary Figure S1).
Pan-Chlamydia PCR, sequencing and data analysis
A pan-Chlamydia PCR was conducted on the tick species in which bacteria belonging to the phylum Chlamydiae had been detected by BLSOM analysis. PCR amplifications were performed using three different sets of primers targeting the 16S rRNA gene (Supplementary Table 1). The successfully amplified products were cloned into a pGEM-T vector (Promega, Madison, WI). Each plasmid clone was sequenced using the BigDye Terminator version 3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and an ABI Prism 3130x genetic analyzer (Applied Biosystems) according to the manufacturer’s instructions. The DNA sequences obtained were submitted to the DDBJ under accession nos. AB725685–AB725705.
The obtained sequences were aligned using ClustalW2 (Larkin et al., 2007) and the resulting alignments were used to generate an uncorrected pairwise distance matrix using the MOTHUR program version 1.25.0 (Schloss et al., 2009). An average neighbour clustering algorithm in the MOTHUR program was used to assign sequences into operational taxonomic units (OTUs), with a 97% sequence similarity cut-off. A representative sequence from each OTU was taxonomically identified using the Classifier tool available on the Ribosomal Database Project (RDP) database (Cole et al., 2009) and was compared against the NCBI GenBank database using a BLASTn search.
Functional annotation and characterization
The metagenomic sequences were annotated using BLASTX (Altschul et al., 1990) against the Clusters of Orthologous Groups (COG) database (Tatusov et al., 1997) with an E-value threshold of 1 × 10−5 and were classified according to COG functional categories. We also identified putative virulence-associated factors using the MvirDB database (Zhou et al., 2007), which consists of sequence information from multiple microbial databases of protein toxins, virulence factors and antibiotic resistance genes. Sequences associated with ‘antibiotic resistance’, ‘pathogenicity islands’ and ‘virulence protein’ were further phylotyped using BLSOM as described above. All sequence reads were used to identify putative virulence-associated factors in BLASTX-based analyses, while only sequences longer than 300 bp were phylotyped.
Results
Kingdom classification
Bacterial/archaeal cells in tick homogenates were purified using centrifugation, DNase treatment and filtration. DNA was extracted from bacteria/archaea-enriched fractions and was subjected to pyrosequencing after whole-genome amplification. The total sequence reads for each species ranged from 21 213 (HFfmn) to 42 258 (AVf), with average lengths between 143.5 bp (IPm) and 391.4 bp (AVm) (Table 2). The sequence reads longer than 300 bp were classified as bacterial/archaeal, eukaryotic, virus, mitochondrial or chloroplast using Kingdom-BLSOM (Figure 2). The percentage of the reads categorized as bacteria/archaea ranged from 37.8% (AVm) to 72.3% (HLfmn), indicating that the present protocol could efficiently enrich bacterial/archaeal cells from tick homogenates. An average of 35% of the eukaryotic reads were clustered together with those of other arthropods, which were included in the Kingdom-BLSOM construction, while the remaining reads were with a variety of eukaryotes including protists and mammals (data not shown).

Kingdom classification of the metagenomic sequences from each tick pool. Sequence reads longer than 300 bp were classified into bacteria/archaea, eukaryotes, viruses, mitochondria or chloroplasts using Kingdom-BLSOM. The percentage of sequences in each category is provided. The pool ID is shown in the centre of each pie chart.
Phylum classification
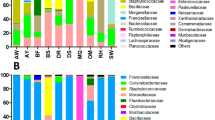
The bacterial/archaeal sequence reads were further classified at the phylum level using Bacteria/Archaea-BLSOM. Between 97.2% (AVf) and 99.4% (HLfmn) of the reads were successfully assigned to specific phyla (Table 2). The number of different phyla obtained from a single library ranged from 20 (HLfmn)–28 (AVm, IOf and IPf). The composition of bacterial/archaeal phyla in each tick pool is shown in Figure 3. Each tick species had a different microbial composition. Differences were also found between males and females within the same tick species. Sequences classified into the phyla Firmicutes and Gammaproteobacteria constituted nearly half of the total sequences in most tick species. Sequences classified into the phylum Alphaproteobacteria were also observed in all tick species, while those classified into the phylum Chlamydiae were observed at high abundance only in IPf, IPm and HFfmn.

Phylum classification of the metagenomic sequences from each tick pool. Sequence reads classified as bacterial/archaeal using Kingdom-BLSOM were further classified at the phylum level using Bacteria/Archaea-BLSOM. Thirty-three phyla are indicated by different colours. The pool ID is shown on the left of the graph.
Genus classification
Between 85.4% (AVm) and 92.3% (ATn) of the sequences characterized into certain bacterial/archaeal phyla were successfully characterized to the genus level using Genus group-BLSOM (Table 2). The number of different genera obtained from a single library ranged from 133 (IRf) to 223 (AVm). The twenty most prevalent bacterial/archaeal genera within each tick species are listed in Table 3, while a complete list of bacterial/archaeal genera recovered from each tick pool is shown in Supplementary Table 2. A total of 64 different genera were detected and some of the dominant bacterial/archaeal genera were commonly observed between tick species. Some of the sequences could not be assigned to known taxa. For example, 4.7% of the sequences in AVm were characterized to the phylum Euryarchaeota, but could not be characterized at the genus level (Table 3). In accordance with the results of phylum classification, many of the dominant bacteria/archaea belonged to genera within the phyla Firmicutes and Gammaproteobacteria. The sequences of known tick-borne pathogens such as Anaplasma, Bartonella, Borrelia, Ehrlichia, Francisella and Rickettsia, as well as those of known tick-symbionts such as Coxiella, Rickettsiella and Wolbachia, were also listed as top 20 genera (Table 3). Only three pools (IPf, IPm and HFfmn) contained bacterial sequences classified into the family Chlamydiaceae (genera Chlamydia and Chlamydophila) as dominant taxa/populations.
Pan-Chlamydia PCR and sequencing analysis
DNA extracted from five tick pools, IPf, IPm, HFf, HFm and HFn, was tested by pan-Chlamydia PCR using three different primer sets. Only one primer set (16S FOR2 and 16S REV2) produced PCR products of the expected size (approximately 260 bp) in all tested samples (data not shown). A total of 14 different clones of partial 16S rRNA gene sequences were recovered, four of which were identified in both tick species (Table 4). The MOTHUR program assigned these 14 sequences into five OTUs, the representative sequences of which were predicted to belong to the genera Neochlamydia, Parachlamydia or Simkania based on RDP database classification. BLASTn analysis of a representative sequence for each OTU showed highest similarity (88%–98%) to Chlamydial 16S rRNA gene sequences recovered from various animal sources including cockroach, sea bass, koala, cat and leafy seadragon (Table 4).
Functional annotation
Functional annotation of the bacterial/archaeal sequences using BLASTX against the COG database indicated similar functional gene compositions between pools, except for AVm (Supplementary Figure 2). More than 80% of the AVm sequences were annotated as ‘Replication, recombination and repair’-related genes. This result may indicate that the sequences in this library were not randomly amplified during the whole-genome amplification process. Sequences associated with putative virulence-associated factors were identified using BLASTX against the MvirDB database. The number of identified sequences differed greatly between libraries: IOm showed the highest number (6715) and ATn the lowest (365) (Figure 4). Most of these were categorized as either ‘antibiotic resistance’, ‘pathogenicity island’ or ‘virulence protein’-related genes. Both IOm and IPf contained a large number of sequences associated with ‘virulence protein’ (n=6515 and 2189, respectively), in comparison to other pools. Sequences associated with ‘pathogenicity island’ were observed to some extent in all pools, with the highest number obtained from IPf (n=2072), followed by IOf, AVm and HLfmn. The origins of these virulence-associated sequences were predicted using Bacteria/Archaea-BLSOM, and five of the most dominant origins of the sequences associated with ‘antibiotic resistance’, ‘pathogenicity island’ and ‘virulence protein’ in each tick pool are shown in Table 5. The most common phyla were Firmicutes, Alphaproteobacteria, Gammaproteobacteria and Chlamydiae, while many of the sequences could not be phylotyped by BLSOM and remained unclassified.

Sequences associated with virulence-associated factors. Sequences associated with putative virulence-associated factors were identified by BLASTX against the MvirDB database (Zhou et al., 2007), applying a cut-off value of 1e-5. The resulting data are shown in seven categories denoted by different colours. The numbers at the bottom of the graph indicate the number of sequences. The numbers in parentheses indicate the percentage of total sequence reads. The pool ID is shown on the left of the graph.
Discussion
The present study investigated the diversity of microbial communities in ticks using pyrosequencing technology coupled with a composition-based data processing approach called BLSOM. In order to provide an overview of microbiomes associated with different tick species, ticks were pooled according to species and subjected to the analyses. We showed that ticks harboured a variety of bacteria, including those previously associated with human/animal diseases such as Anaplasma, Bartonella, Borrelia, Ehrlichia, Francisella and Rickettsia, as well as potential pathogens such as Chlamydia. This is a first attempt to apply BLSOM to the detection of potential pathogens. Since this approach can be directly applied to other vector arthropods of medical and veterinary importance, it has great potential for the detection of diverse microbes, including as-yet unidentified microorganisms, and to pre-empt emerging infectious diseases.
Unlike the conventional sequence homology searches used in most metagenomic studies, BLSOM does not require orthologous sequences for phylogenetic classification of metagenomic sequences. This is a great advantage, especially when applied to a group composed of poorly characterized microorganisms (Abe et al., 2005). In addition, ticks have highly variable genome sizes, ranging from nearly one third to over two times the size of the human genome, but only one whole-genome sequencing project is underway (Nene, 2009). This indicates that alignment-based techniques may encounter difficulties in sorting out tick-derived sequences in the metagenomic libraries. On the other hand, BLSOM can separate bacterial/archaeal sequences from those of eukaryotes with high accuracy (Supplementary Figure S1) and thus have great potential for detecting symbiotic and commensal microorganisms in eukaryotic hosts with scarce or no genomic information. Despite the fact that Kingdom-BLSOM did not include tick genome sequences as these were not available at the time of construction, an average of 35% of the eukaryotic sequences were clustered together with those of other arthropods and thus likely to be derived from tick cells. Once it becomes available, the inclusion of a tick genome in BLSOM construction would facilitate the identification of eukaryotic reads as being tick-derived.
In a previous pyrosequencing study, in which the whole bodies of I. ricinus were directly subjected to DNA and RNA extraction, only 0.095% of the genomic DNA and 0.30% of the cDNA-derived sequences were assigned to known bacterial taxa (Carpi et al., 2011). Although this shotgun strategy seems ideal for capturing a snapshot of a microbial community with minimum bias introduced during sample preparation, there is a concern that genomic data for bacteria/archaea present in very low numbers may be masked by the huge amount of host-genome sequences. To address this, our strategy included a process of microbial enrichment using centrifugation, DNase treatment and filtration, to enrich bacterial/archaeal cells from tick homogenates prior to pyrosequencing. The microbial profiles obtained in this study, therefore, may not completely reflect the whole picture regarding microbial composition in the tested ticks due to the biases introduced during the microbial enrichment and whole-genome amplification processes, especially in the case of AVm, where the sequences in the library were not likely to be randomly amplified (Supplementary Figure 2). Nonetheless, the fact that nearly half of the sequence reads were identified as bacterial/archaeal, and were associated with a diverse range of bacterial/archaeal phyla, indicated sufficient coverage by our method to obtain an overview of tick microbiomes.
Sequences of genera containing several tick-borne pathogens, such as Anaplasma, Borrelia and Rickettsia, were present in the tested samples (Table 3 and Supplementary Table 2). Further identification to the species level can be achieved by the use of traditional methods such as conventional species-specific PCR. This can be done to confirm the presence of tick-borne bacterial species such as A. phagocytophilum, B. burgdorferi s.l., R. japonica and R. tamurae, which have previously been detected in the tested tick species (Morita et al., 1990; Miyamoto et al., 1992; Hofhuis et al., 2006; Murase et al., 2011). The data obtained in the present study, however, are useful for broadening our understanding of phylogenetic diversity among bacterial genera, and the extent of their habitats. For example, the genera Chlamydia and Chlamydophila were predicted to be harboured by IPf, IPm and HFfmn in high abundance (Table 3). Further pan-Chlamydia PCR and sequencing analysis supported the existence of microorganisms possibly related to the genera Neochlamydia, Parachlamydia and Simkania (Table 4). Some representatives of these genera are implicated in diseases affecting their host animals (von Bomhard et al., 2003; Meijer et al., 2006; Corsaro et al., 2007). Since only the sequences of the genera Chlamydia and Chlamydophila were available from the phylum Chlamydiae at the time of BLSOM construction, those of the genera Neochlamydia, Parachlamydia and Simkania were not included in Genus group-BLSOM, so sequences from the phylum Chlamydiae were allocated to either genus Chlamydia or Chlamydophila. This technical limitation will be overcome as sequence data from a wider range of microorganisms become available. Although possible transmission of Chlamydial bacteria to cattle and humans through ticks was reported in several previous studies (Caldwell and Belden, 1973; McKercher et al., 1980; Facco et al., 1992), there has been no molecular genetic evidence to support these observational studies. Thus, this is the first molecular genetic evidence supporting the presence of Chlamydial organisms in ticks. Given the wide genetic diversity and host range, including several arthropods, of members of this phylum of bacteria (Horn, 2008), further research is warranted to improve the understanding of their biology, especially with respect to their pathogenic potential for humans and animals.
Genus group-BLSOM identified 133–223 different genera from each single tick pool (Table 2). The presence of a variety of bacterial/archaeal genera in ticks was demonstrated in previous metagenomic studies; in R. microplus and I. ricinus, 120 and 108 different genera were identified, respectively, using a rDNA-based amplicon pyrosequencing approach (Andreotti et al., 2011; Carpi et al., 2011). This discrepancy may be explained by the fact that these two approaches rely on different analytic principles. One possible explanation is that BLSOM might have misclassified genome segments introduced through horizontal gene transfer (HGT) (Abe et al., 2005). Tamames and Moya (2008) reported evidence of HGT in 0.8%–1.5% and 2%–8% of the sequences in environmental metagenomic libraries using phylogenetic and composition-based methods, respectively, indicating that the present approach may require further refinement; for example, by combining BLSOM with rDNA-based phylogenetic classification as proposed elsewhere (Abe et al., 2005). It is also possible that the Genus group-BLSOM has overestimated the microbial diversity in the tested samples as a result of a decreased classification specificity on the genus level (Supplementary Figure S1). This drawback can be remedied by using only longer sequences to improve the specificity of the analysis or by using more genome sequences associated with the habitat under study during BLSOM construction (Abe et al., 2005; Weber et al., 2011).
The present study included the ticks maintained under laboratory conditions. The microbial diversity of those ticks was comparable to that of those collected in the field in terms of the number of different genus identified (Table 2). Environmental factors and developmental stages were reported to affect microbial communities in ticks (Clay et al., 2008; van Overbeek et al., 2008; Andreotti et al., 2011; Carpi et al., 2011). Therefore, a further comparison of microbial diversity between laboratory colony and field ticks of the same species may be useful to elucidate environmental factors playing key roles on microbial compositions. IO and IP were sampled at the same site and time and have a common host preference for sika deer (Cervus nippon) in the sampling area (Isogai et al., 1996), allowing a comparison of microbial compositions between these two tick species. IO harboured Rickettsia and Ehrlichia, but IP harboured Bartonella, at high abundance, despite the presence of Borrelia and Francisella in both tick species at similar abundance (Table 3). These findings may be useful for improving the understanding of the vector potential of different tick species. In addition to these species differences, microbial communities are expected to vary between individual ticks as reported elsewhere (Andreotti et al., 2011; Carpi et al., 2011). One aspect to be addressed, which may help to explain these differences, is the interaction between microbial lineages. Indeed, antagonistic interactions between microbial lineages in ticks, including pathogenic Rickettsia, were demonstrated previously (Macaluso et al., 2002; de la Fuente et al., 2003). Thus, one of the most interesting challenges for future studies on tick microbiomes might be the association between microbial composition in ticks and the potential for pathogen transmission. The pictures of whole microbiomes harboured by each tick species generated in the present study provide the foundation for a statistical correlation analysis between pathogens and certain microbial lineages in individual ticks. Such studies could lead to the identification of microbiological factors affecting the prevalence and persistence of pathogenic lineages in ticks.
The sequences associated with putative virulence-associated factors were identified from metagenomic libraries using BLASTX-based methods (Figure 4). It should be noted, however, that some of the factors, such as type III secretion systems, are responsible for both bacterial pathogenesis and symbiosis, including mutualism and commensalism (Coombes, 2009), and thus not all factors are directly or necessarily related to human/animal pathogens. The present study predominantly identified sequences related to antibiotic resistance (Figure 4). It has been recognized for some time that antibiotic resistance genes are not only associated with pathogens but also with a wide variety of environmental microbes (Allen et al., 2010). Several lines of evidence indicate that some of the clinically relevant resistance mechanisms in pathogenic organisms originated from such environmental bacteria through HGT (Wright, 2010). Direct contact between pathogens and resistance genes in commensals harboured by ticks could therefore represent a mechanism for the emergence of resistant pathogens.
Several studies described mutualistic relationships between microbes and host arthropods (Currie et al., 1999; Kaltenpoth et al., 2005; Scott et al., 2008). It is plausible that the tick microbiome also affects aspects of tick physiology. This was for example demonstrated by a reduction in reproductive fitness of Amblyomma americanum females following antibiotic treatment, suggesting that the microbe plays a role in tick growth and development (Zhong et al., 2007). Further studies should consider the potential contributions of tick microbiomes to the survival of ticks in the environment; this could provide novel targets for the control of ticks and tick-borne pathogens.
BLSOM was used to estimate the phylogenetic origins of the sequences associated with putative virulence-associated factors; however, many of these remained unclassified (Table 5). This may indicate that those sequences had retained their functions, but lost sequence characteristics such as oligonucleotide frequency. One possible explanation is that the unclassified sequences were introduced through ancient HGT, and lost their oligonucleotide frequency in the recipient genome (either tick or other microbes). Indeed, the oligonucleotide composition of sequences introduced through ancient HGT drift over time (Brown, 2003), makes it difficult to correctly phylotype those sequences using composition-based methods such as BLSOM. Furthermore, numerous reports have associated bacterial virulence factors with HGT (Pallen and Wren, 2007; Ho Sui et al., 2009; Juhas et al., 2009). Another explanation is that these sequences simply originated from as-yet unclassified microorganisms. Regardless, the detection of virulence-associated factors in ticks may suggest either the existence or possible emergence of tick-borne pathogens and, thus, warrants further investigation to assess potential risks to human and animal health.
In conclusion, these results provide a foundation for the construction of a database of tick microbes, which may aid in the prediction of emerging tick-borne diseases. The present approach can be extended to detect or predict emerging pathogens in other vector arthropods such as mosquitoes and flies. A comprehensive understanding of tick microbiomes will also be useful for understanding tick biology, including vector competence and interactions with pathogens.
References
Abe T, Kanaya S, Kinouchi M, Ichiba Y, Kozuki T, Ikemura T . (2003). Informatics for unveiling hidden genome signatures. Genome Res 13: 693–702.
Abe T, Sugawara H, Kinouchi M, Kanaya S, Ikemura T . (2005). Novel phylogenetic studies of genomic sequence fragments derived from uncultured microbe mixtures in environmental and clinical samples. DNA Res 12: 281–290.
Allen HK, Donato J, Wang HH, Cloud-Hansen KA, Davies J, Handelsman J . (2010). Call of the wild: antibiotic resistance genes in natural environments. Nat Rev Microbiol 8: 251–259.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ . (1990). Basic local alignment search tool. J Mol Biol 215: 403–410.
Andreotti R, Pérez de León AA, Dowd SE, Guerrero FD, Bendele KG, Scoles GA . (2011). Assessment of bacterial diversity in the cattle tick Rhipicephalus (Boophilus) microplus through tag-encoded pyrosequencing. BMC Microbiol 11: 6.
Bowman AS, Nuttall PA . (2008) Ticks: Biology, Disease and Control. Cambridge University Press: Cambridge.
Benson MJ, Gawronski JD, Eveleigh DE, Benson DR . (2004). Intracellular symbionts and other bacteria associated with deer ticks (Ixodes scapularis) from Nantucket and Wellfleet, Cape Cod, Massachusetts. Appl Environ Microbiol 70: 616–620.
Beugnet F, Marié JL . (2009). Emerging arthropod-borne diseases of companion animals in Europe. Vet Parasitol 163: 298–305.
Brown JR . (2003). Ancient horizontal gene transfer. Nat Rev Genet 4: 121–132.
Caldwell HD, Belden EL . (1973). Studies of the role of Dermacentor occidentalis in the transmission of bovine chlamydial abortion. Infect Immun 7: 147–151.
Carpi G, Cagnacci F, Wittekindt NE, Zhao F, Qi J, Tomsho LP et al. (2011). Metagenomic profile of the bacterial communities associated with Ixodes ricinus ticks. PLoS One 6: e25604.
Chan C-KK, Hsu AL, Tang S-L, Halgamuge SK . (2008). Using growing self-organising maps to improve the binning process in environmental whole-genome shotgun sequencing. J Biomed Biotechnol 2008: doi:10.1155/2008/513701.
Clay K, Klyachko O, Grindle N, Civitello D, Oleske D, Fuqua C . (2008). Microbial communities and interactions in the lone star tick, Amblyomma americanum. Mol Ecol 17: 4371–4381.
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ et al. (2009). The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37: D141–D145.
Coombes BK . (2009). Type III secretion systems in symbiotic adaptation of pathogenic and non-pathogenic bacteria. Trends Microbiol 17: 89–94.
Corsaro D, Thomas V, Goy G, Venditti D, Radek R, Greub G . (2007). ‘Candidatus Rhabdochlamydia crassificans’, an intracellular bacterial pathogen of the cockroach Blatta orientalis (Insecta: Blattodea). Syst Appl Microbiol 30: 221–228.
Cox-Foster DL, Conlan S, Holmes EC, Palacios G, Evans JD, Moran NA et al. (2007). A metagenomic survey of microbes in honey bee colony collapse disorder. Science 318: 283–287.
Currie CR, Mueller UG, Malloch D . (1999). The agricultural pathology of ant fungus gardens. Proc Natl Acad Sci USA 96: 7998–8002.
de la Fuente J, Blouin EF, Kocan KM . (2003). Infection exclusion of the rickettsial pathogen Anaplasma marginale in the tick vector Dermacentor variabilis. Clin Diagn Lab Immunol 10: 182–184.
Dick GJ, Andersson AF, Baker BJ, Simmons SL, Thomas BC, Yelton AP et al. (2009). Community-wide analysis of microbial genome sequence signatures. Genome Biol 10: R85.
Ezaki T, Suzuki S . (1982). Achromopeptidase for lysis of anaerobic gram-positive cocci. J Clin Microbiol 16: 844–846.
Facco F, Grazi G, Bonassi S, Magnani M, Di Pietro P . (1992). Chlamydial and rickettsial transmission through tick bite in children. Lancet 339: 992–993.
Grenier AM, Duport G, Pagès S, Condemine G, Rahbé Y . (2006). The phytopathogen Dickeya dadantii (Erwinia chrysanthemi 3937) is a pathogen of the pea aphid. Appl Environ Microbiol 72: 1956–1965.
Ho Sui SJ, Fedynak A, Hsiao WW, Langille MG, Brinkman FS . (2009). The association of virulence factors with genomic islands. PLoS One 4: e8094.
Hofhuis A, van der Giessen JWB, Borgsteede FHM, Wielinga PR, Notermans DW, van Pelt W . (2006). Lyme borreliosis in the Netherlands: strong increase in GP consultations and hospital admissions in past 10 years. Euro Surveillance: European Communicable Disease Bulletin 11: E060622.2.
Horn M . (2008). Chlamydiae as symbionts in eukaryotes. Annu Rev Microbiol 62: 113–131.
Imaoka K, Kaneko S, Tabara K, Kusatake K, Morita E . (2011). The First Human Case of Rickettsia tamurae Infection in Japan. Case Rep Dermatol 3: 68–73.
Isogai E, Isogai H, Masuzawa T, Postic D, Baranton G, Kamewaka Y et al. (1996). Borrelia burgdorferi sensu lato in an endemic environment: wild sika deer (Cervus nippon yesoensis) with infected ticks and antibodies. Microbiol Immunol 40: 13–19.
Juhas M, van der Meer JR, Gaillard M, Harding RM, Hood DW, Crook DW . (2009). Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev 33: 376–393.
Kaltenpoth M, Göttler W, Herzner G, Strohm E . (2005). Symbiotic bacteria protect wasp larvae from fungal infestation. Curr Biol 15: 475–479.
Kanaya S, Kinouchi M, Abe T, Kudo Y, Yamada Y, Nishi T et al. (2001). Analysis of codon usage diversity of bacterial genes with a self-organizing map (SOM): characterization of horizontally transferred genes with emphasis on the E. coli O157 genome. Gene 276: 89–99.
Kohonen T . (1990). The self-organizing map. Proceedings of the IEEE pp 1464–1480.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948.
Lewis D . (1979). The detection of rickettsia-like microorganisms within the ovaries of female Ixodes ricinus ticks. Z Parasitenkd 59: 295–298.
Macaluso KR, Sonenshine DE, Ceraul SM, Azad AF . (2002). Rickettsial infection in Dermacentor variabilis (Acari: Ixodidae) inhibits transovarial transmission of a second Rickettsia. J Med Entomol 39: 809–813.
Martin C, Diaz NN, Ontrup J, Nattkemper TW . (2008). Hyperbolic SOM-based clustering of DNA fragment features for taxonomic visualization and classification. Bioinformatics 24: 1568–1574.
Mavromatis K, Ivanova N, Barry K, Shapio H, Goltsman E, McHardy AC et al. (2007). Use of simulated data sets to evaluate the fidelity of metagenomic processing methods. Nat Methods 4: 495–500.
McKercher DG, Wada EM, Ault SK, Theis JH . (1980). Preliminary studies on transmission of Chlamydia to cattle by ticks (Ornithodoros coriaceus). Am J Vet Res 41: 922–924.
Meijer A, Roholl PJ, Ossewaarde JM, Jones B, Nowak BF . (2006). Molecular evidence for association of chlamydiales bacteria with epitheliocystis in leafy seadragon (Phycodurus eques), silver perch (Bidyanus bidyanus), and barramundi (Lates calcarifer). Appl Environ Microbiol 72: 284–290.
Miyamoto K, Nakao M, Uchikawa K, Fujita H . (1992). Prevalence of Lyme borreliosis spirochetes in ixodid ticks of Japan, with special reference to a new potential vector, Ixodes ovatus (Acari: Ixodidae). J Med Entomol 29: 216–220.
Morita C, Yamamoto S, Tsuchiya K, Yoshida Y, Yabe T, Kawabata N et al. (1990). Prevalence of spotted fever group rickettsia antibody in Apodemus speciosus captured in an endemic focus in Miyazaki Prefecture, Japan. Jpn J Med Sci Biol 43: 15–18.
Murase Y, Konnai S, Hidano A, Githaka NW, Ito T, Takano A et al. (2011). Molecular detection of Anaplasma phagocytophilum in cattle and Ixodes persulcatus ticks. Vet Microbiol 149: 504–507.
Nakabachi A, Ishikawa H . (1999). Provision of riboflavin to the host aphid, Acyrthosiphon pisum, by endosymbiotic bacteria,. Buchnera. J Insect Physiol 45: 1–6.
Nene V . (2009). Tick genomics--coming of age. Front Biosci 14: 2666–2673.
Noda H, Munderloh UG, Kurtti TJ . (1997). Endosymbionts of ticks and their relationship to Wolbachia spp. and tick-borne pathogens of humans and animals. Appl Environ Microbiol 63: 3926–3932.
Pallen MJ, Wren BW . (2007). Bacterial pathogenomics. Nature 449: 835–842.
Parola P . (2004). Tick-borne rickettsial diseases: emerging risks in Europe. Comp Immunol Microbiol Infect Dis 27: 297–304.
Parola P, Davoust B, Raoult D . (2005). Tick- and flea-borne rickettsial emerging zoonoses. Vet Res 36: 469–492.
Roshdy MA . (1968). A rickettsialike microorganism in the tick Ornithodoros savignyi: Observations on its structure and distribution in the tissues of the tick. J Invertebr Pathol 11: 155–169.
Scarborough CL, Ferrari J, Godfray HC . (2005). Aphid protected from pathogen by endosymbiont. Science 310: 1781.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541.
Scott JJ, Oh DC, Yuceer MC, Klepzig KD, Clardy J, Currie CR . (2008). Bacterial protection of beetle-fungus mutualism. Science 322: 63.
Spolidorio MG, Labruna MB, Mantovani E, Brandao PE, Richtzenhain LJ, Yoshinari NH . (2010). Novel spotted fever group rickettsiosis, Brazil. Emerg Infect Dis 16: 521–523.
Suen G, Scott JJ, Aylward FO, Adams SM, Tringe SG, Pinto-Tomás AA et al. (2010). An insect herbivore microbiome with high plant biomass-degrading capacity. PLoS Genet 6: e1001129.
Tamames J, Moya A . (2008). Estimating the extent of horizontal gene transfer in metagenomic sequences. BMC Genomics 9: 136.
Tatusov RL, Koonin EV, Lipman DJ . (1997). A genomic perspective on protein families. Science 278: 631–637.
Tsuchida T, Koga R, Horikawa M, Tsunoda T, Maoka T, Matsumoto S et al. (2010). Symbiotic bacterium modifies aphid body color. Science 330: 1102–1104.
Uehara H, Iwasaki Y, Wada C, Ikemura T, Abe T . (2011). A novel bioinformatics strategy for searching industrially useful genome resources from metagenomic sequence libraries. Genes Genet Syst 86: 53–66.
van Overbeek L, Gassner F, van der Plas CL, Kastelein P, Nunes-da Rocha U, Takken W . (2008). Diversity of Ixodes ricinus tick-associated bacterial communities from different forests. FEMS Microbiol Ecol 66: 72–84.
von Bomhard W, Polkinghorne A, Lu ZH, Vaughan L, Vögtlin A, Zimmermann DR et al. (2003). Detection of novel chlamydiae in cats with ocular disease. Am J Vet Res 64: 1421–1428.
Warnecke F, Luginbühl P, Ivanova N, Ghassemian M, Richardson TH, Stege JT et al. (2007). Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450: 560–565.
Weber M, Teeling H, Huang S, Waldmann J, Kassabgy M, Fuchs BM et al. (2011). Practical application of self-organizing maps to interrelate biodiversity and functional data in NGS-based metagenomics. ISME J 5: 918–928.
Weiss B, Aksoy S . (2011). Microbiome influences on insect host vector competence. Trends Parasitol 27: 514–522.
Wright GD . (2010). Antibiotic resistance in the environment: a link to the clinic? Curr Opin Microbiol 13: 589–594.
Yu XJ, Liang MF, Zhang SY, Liu Y, Li JD, Sun YL et al. (2011). Fever with thrombocytopenia associated with a novel bunyavirus in China. N Engl J Med 364: 1523–1532.
Zhong J, Jasinskas A, Barbour AG . (2007). Antibiotic treatment of the tick vector Amblyomma americanum reduced reproductive fitness. PLoS One 2: e405.
Zhou CE, Smith J, Lam M, Zemla A, Dyer MD, Slezak T . (2007). MvirDB--a microbial database of protein toxins, virulence factors and antibiotic resistance genes for bio-defence applications. Nucleic Acids Res 35: D391–D394.
Acknowledgements
We thank everyone who helped with tick collection, especially Nariaki Nonaka, Jacqueline Schipper, Joseph W. Magona, Joseph M. Kamau, Noboru Sasaki and Noriko Nakao. The first author was supported by a research grant fellowship from the Japanese Society for the Promotion of Science (JSPS) for young scientists. This work was supported by a Grant-in-Aid for JSPS fellows and for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT), the program of Funding Research Center for Emerging and Re-emerging Infectious Disease, MEXT. The BLSOM calculation was done, in part, by the Earth Simulator of the Japan Agency for Marine-Earth Science and Technology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Rights and permissions
About this article
Cite this article
Nakao, R., Abe, T., Nijhof, A. et al. A novel approach, based on BLSOMs (Batch Learning Self-Organizing Maps), to the microbiome analysis of ticks. ISME J 7, 1003–1015 (2013). https://doi.org/10.1038/ismej.2012.171
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2012.171
Keywords
This article is cited by
-
Cross-alteration of murine skin and tick microbiome concomitant with pathogen transmission after Ixodes ricinus bite
Microbiome (2023)
-
Characterization of the bacterial microbiome of Swedish ticks through 16S rRNA amplicon sequencing of whole ticks and of individual tick organs
Parasites & Vectors (2023)
-
Characterization of the bacterial microbiome of Amblyomma scalpturatum and Amblyomma ovale collected from Tapirus terrestris and Amblyomma sabanerae collected from Chelonoidis denticulata, Madre de Dios- Peru
BMC Microbiology (2022)
-
The Microbiota of Ixodes ricinus and Dermacentor reticulatus Ticks Collected from a Highly Populated City of Eastern Europe
Microbial Ecology (2022)
-
Temporal patterns in Ixodes ricinus microbial communities: an insight into tick-borne microbe interactions
Microbiome (2021)
