
Abstract
The addition of organic compounds to groundwater in order to promote bioremediation may represent a new selective pressure on subsurface microorganisms. The ability of Geobacter sulfurreducens, which serves as a model for the Geobacter species that are important in various types of anaerobic groundwater bioremediation, to adapt for rapid metabolism of lactate, a common bioremediation amendment, was evaluated. Serial transfer of five parallel cultures in a medium with lactate as the sole electron donor yielded five strains that could metabolize lactate faster than the wild-type strain. Genome sequencing revealed that all five strains had non-synonymous single-nucleotide polymorphisms in the same gene, GSU0514, a putative transcriptional regulator. Introducing the single-base-pair mutation from one of the five strains into the wild-type strain conferred rapid growth on lactate. This strain and the five adaptively evolved strains had four to eight-fold higher transcript abundance than wild-type cells for genes for the two subunits of succinyl-CoA synthase, an enzyme required for growth on lactate. DNA-binding assays demonstrated that the protein encoded by GSU0514 bound to the putative promoter of the succinyl-CoA synthase operon. The binding sequence was not apparent elsewhere in the genome. These results demonstrate that a single-base-pair mutation in a transcriptional regulator can have a significant impact on the capacity for substrate utilization and suggest that adaptive evolution should be considered as a potential response of microorganisms to environmental change(s) imposed during bioremediation.
Similar content being viewed by others

Introduction
The unnatural conditions that some strategies for bioremediation impose on subsurface microbial communities may provide sufficient selective pressure for the evolution of new microbial strains that are better adapted to the engineered environment. For example, a strategy for bioremediation of uranium-contaminated groundwater is to accelerate the growth of Geobacter species with the addition of acetate to promote reductive precipitation of uranium (Anderson et al., 2003; Chang et al., 2005; N’Guessan et al., 2008). Studies with Geobacter sulfurreducens demonstrated that selective pressure for faster growth on Fe(III) oxides, the primary electron acceptor supporting Geobacter growth during uranium bioremediation (Finneran et al., 2002), resulted in replicate strains, which accumulated mutations that increased the production of microbial pili (Reguera et al., 2005, 2006) and other proteins that enhance the rate of Fe(III) oxide reduction (Tremblay et al., 2011).
In addition to the demand for faster growth, another artificial demand on subsurface communities during bioremediation can be for rapid utilization of electron donors that are not commonly abundant in the subsurface. For example, lactate is a convenient electron donor for promoting anaerobic metabolism because it can be provided from one-time emplacements of slow-release formulations (Hazen and Tabak, 2005). In contrast to acetate, which is a major intermediate in carbon and electron flow in the subsurface, lactate is expected to be a minor constituent under conditions present during the stimulation of bioremediation in the subsurface (Lovley and Chapelle, 1995). Therefore, a consequence of adding high concentrations of this usually rare substrate may be an adaptive response to utilize lactate more effectively.
The ability of laboratory cultures of Escherichia coli to adapt to more effectively metabolize substrates under selective pressure is well documented (Ferea et al., 1999; Shaver et al., 2002; Wick et al., 2002; Cooper et al., 2003; Herring et al., 2006). The availability of technology to rapidly identify mutations in adapted strains, and then evaluate the contributions of those mutations to adaptation with genetic manipulation, has greatly accelerated the understanding of the mechanisms for adaptation to new substrates (Brockhurst et al., 2010). Genomic changes promoting adaptation to new substrates have included mutations that enhance the kinetics of enzymes acting on the substrate (Fong et al., 2005a, 2005b; Herring et al., 2006; Conrad et al., 2009; Lee and Palsson, 2010) and mutations in promoter regions and global regulatory elements (Treves et al., 1998; Herring et al., 2006; Pelosi et al., 2006); as well as RNA polymerases (Herring et al., 2006; Conrad et al., 2010). Surprisingly, there have been fewer reports of adaptation to the use of a new substrate via mutations in known transcriptional regulators (Philippe et al., 2007; Barrick et al., 2009) despite the fact that changes in transcriptional regulators are thought to have key roles in the evolution of new microbial species (Dekel and Alon, 2005; Babu and Aravind, 2006; Babu et al., 2007), a concept further supported with the observation that transcriptional regulatory networks evolve and change faster than other collaborative biological networks such as protein interaction networks and metabolic pathway networks (Shou et al., 2011).
G. sulfurreducens, which has been used extensively as a model for the Geobacter species that predominate during groundwater bioremediation (Lovley, 2003), was previously reported to be unable to grow on lactate (Caccavo et al., 1994). Here we report that when G. sulfurreducens was provided with lactate as the sole electron donor it adapted over time to grow effectively on lactate. Unlike many other previous studies on adaptive evolution for expanded substrate utilization in other organisms, the adaptation in G. sulfurreducens could be attributed to mutations in a previously unstudied transcriptional regulator.
Materials and methods
Experimental evolution and growth conditions
The ancestral strain for the adaptive evolution experiments was from a frozen stock of G. sulfurreducens PCA genome strain ATCC# 51573 (Methe et al., 2003). The growth medium was the previously described NBAF medium (Coppi et al., 2001), with the modification that 10 mM lactic acid was substituted for 15 mM acetate as the sole electron donor/carbon source. A total of 40 mM fumarate was provided as the sole electron acceptor. Media were dispensed into pressure tubes, bubbled with an 80:20 nitrogen:carbon dioxide gas mixture to remove dissolved oxygen and obtain a final pH of 6.9–7.0 and then sealed with butyl rubber stoppers. All media were sterilized by autoclaving and all incubations were at 30 °C. Standard anaerobic techniques were used throughout (Balch et al., 1979). Five cultures were grown in parallel. Growth was measured by optical density at a wavelength of 600 nm with a Spectronic Genesys 2 spectrophotometer (Thermo Electron Corporation, Waltham, MA, USA) equipped with culture tube reading apparatus. The parallel cultures were grown until an absorbance of 0.4 was reached, approximately mid-late logarithmic growth phase and were serially transferred always at this point by means of a 1% (0.1 ml) transfer to the next tube. The maximum absorbance of the cultures once they had reached stationary phase was ∼0.5, and the maximum absorbance did not change over the course of the laboratory evolution experiment.
Resequencing of evolved strain
Cells were collected by centrifugation and genomic DNA was extracted using the Epicenter Master Pure DNA Purification kit (Epicentre Biotechnologies, Madison, WI, USA) following the manufacturer's instructions. Sequencing of the evolved genomes using Illumina sequencing, and genome assembly was performed as described previously (Tremblay et al., 2011). All mutations reported by resequencing were confirmed in-house by PCR amplification of the sequence and Sanger sequencing of the DNA fragment using an Applied Biosystems (Life Technologies Corporation, Carlsbad, CA, USA) 3730xl DNA Analyzer.
Mutation knock-in
To determine if a single-point mutation could account for the phenotype of a lactate-adapted strain, a copy of the mutated gene was introduced into the genome with the Cre-Lox system (Marx and Lidstrom, 2002). First the GSU0514 gene was replaced with a kanamycin-resistance gene such that the coding region from amino acid residues 36Leu to 252Ile was deleted. To achieve the deletion strain, double-crossover homologous recombination was carried out by electroporation (Coppi et al., 2001) with the linear DNA fragment consisting of the kanamycin-resistance gene flanked by 0.7 kbp DNA fragments containing the upstream and the downstream regions of the GSU0514 gene. These flanking DNA fragments were amplified by PCR with primers, 5′-TCTCTCGAGCAAATTTAACGAGTTCCTC-3′ and 5′-TCAAGCTTCGGTCACGCCCAGCTCGTC-3′ (XhoI and HindIII sites are underlined) and, 5′-TCGAATTCCATGAAGCTCGGCTTTCA-3′ and 5′-TCTCTAGACACCGGCAACCGACTGTGAA-3′ (EcoRI and XbaI sites are underlined), respectively. The DNA fragment of the kanamycin-resistance gene was amplified by PCR with primers, 5′-GCATGAGAATTCCTGACGGAACAGCGGGAAGTCCAGC-3′ and 5′-GCTATGAAGCTTTCATAGAAGGCGGCGGTGGAATCGAA-3′ (EcoRI and HindIII sites are underlined), and pBBR1MCS-2 (Kovach et al., 1994) as a template. These PCR products were digested with restriction enzymes, ligated and cloned in the plasmid. The sequences of these cloned fragments were confirmed by DNA sequencing. After electroporation, kanamycin-resistant transformants were isolated and inoculated in NBAF medium supplemented with 200 μg ml−1 kanamycin. The replacement was confirmed by PCR amplification. This leaves the targeted gene inactive, and grants antibiotic resistance to the cell.
GSU0514 and its flanking regions were amplified by PCR using the primers 5′-CAAATTTAACGAGTTCCTC-3′, and 5′-CACCGGCAACCGACTGTGAA-3′ from genomic DNA extracted from the adapted strain, ZMS123, containing the single-nucleotide polymorphism (SNP). The fragment was cloned into the pCR2.1 TOPO vector (Invitrogen, Life Technologies Corporation, Carlsbad, CA, USA), creating the plasmid pZS. The plasmid was digested with NheI, gel purified and used to clone the XbaI fragment containing a gentamicin resistance cassette flanked with two LoxP sites isolated from the plasmid pUC::GmRLoxP (Aklujkar and Lovley, 2010). The plasmid (pWA4) thus constructed was digested with EcoRI and the 2.3-kbp fragment was gel purified. Then, the purified fragment was used to transform electrocompetent cells of the GSU0514 knock-out strain as described previously (Coppi et al., 2001). After electroporation, the cells were recovered for 18 h in the lactate growth medium supplemented with cysteine (1 mM) and 0.1% yeast extract. Transformants were selected on agar-solidified lactate growth medium containing 20 μg ml−1 gentamicin. Single colonies were inoculated into a liquid medium containing 200 μg ml−1 kanamycin to test for the loss of the kanamycin-resistance gene. Kanamycin-sensitive colonies were checked by PCR to confirm the insertion of the mutagenic fragment into the chromosome. Locus specific primers (GSU0514F, 5′-TGTCTCCTCCTGAAAATAGAAC-3′; GSU0514R, 5′-GGGCTTTTCTGCTAGCGATGAAAGCCGAGCTTCATG-3′) were used for the PCR amplification under the following conditions: 94 °C for 1 min followed by 30 cycles of 94 °C for 30 s; 65 °C for 45 s; 72 °C for 90 s and finally 72 °C for 10 min. To evict the gentamicin cassette, electrocompetent cells of the knock-in mutant were transformed with pCM158 expressing the Cre-recombinase and kanamycin-resistance genes (Marx and Lidstrom, 2002). After electroporation, transformants were recovered overnight in a liquid medium amended with cysteine and yeast extract. Cells from the recovery culture were plated onto a medium with 200 μg ml−1 kanamycin. Kanamycin-resistant mutants were purified twice non-selectively without antibiotics and then tested for the loss of the Kanr and Gmr cassettes. Mutations were confirmed by Sanger sequencing, and LA Taq DNA polymerase from Takara (Takara Bio Inc., Otsu, Japan) was used for the PCR.
qRT-PCR analysis
Triplicate 100 ml bottles were inoculated with a 1% transfer. Wild-type G. sulfurreducens, ZMS123 and the single-base-pair knock-in strain were grown on NBLF (10 mM lactate, 40 mM fumarate) media. All cultures were harvested in mid-log phase, when they reached an optical density of 0.35 as determined by a spectrophotometer at a wavelength of 600 nm. The cultures were each split into two 50 ml aliquots, spun down at 4 °C at >3000 g for 15 min. The cell pellets were then flash frozen with liquid N2 and stored at −80 °C. RNA was extracted from the triplicate cultures using the Qiagen RNeasy Midi Kit (Qiagen Inc., Valencia, CA, USA). The extracted RNA was then treated with the Ambion DNA-free Kit (Life Technologies Corporation, Carlsbad, CA, USA) so as to remove all genomic DNA from the sample. A control PCR reaction was run with G. sulfurreducens specific primers to confirm that no genomic DNA remained in the tube after the DNAse treatment. The concentration was determined by absorbance at 260/280 using a NanoDrop ND-1000 spectrophotometer (Nanodrop Products, Wilmington, DE, USA). QPCR was performed as described previously (Holmes et al., 2009) on the ABI Prism 7900HT Sequence Detection System (Applied Biosystems) using primers specific for the succinyl CoA synthetase α-subunit, GSU1058 (sucC), 5′-ACCTCTCGCTACTGCCTCAT-3′ and 5′-TCGATGGTGAGAACATGGAT-3′ and the succinyl CoA synthetase β-subunit, GSU1059 (sucD), 5′-GGTCTCTTCCATACCCAGCA-3′ and 5′-TTGAAGACCGGTATCCCTTC-3′. The relative abundance of transcript levels of GSU1058 and GSU1059 from all five replicate adapted strains was compared with that of the wild-type G. sulfurreducens grown on a lactate-fumarate medium.
Construction of GSU0514 expression vector and purification of GSU0514
The DNA fragment encoding GSU0514 was amplified from wild-type G. sulfurreducens genomic DNA by PCR with primers 5′-TCTCATATGGCGAAGAAGGACAAATCCGAA-3′ and 5′-TCTCTCGAGCCGATGAAAGCCGAGCTTCATGGATA-3′ (NdeI and XhoI sites are underlined). The PCR products were cloned into pET24b (Novagen, EMB Chemicals, Merck, Darmstadt, Germany). The sequence of the PCR products was confirmed. GSU0514 was prepared as histidine-tagged protein at the C-terminus. Overexpression of GSU0514 was achieved by Autoinduction system (Novagen) as instructed by the manufacturer. Purification of GSU0514 was performed as described previously (Ueki and Inouye, 2002).
DNA-binding assay
Promoter regions upstream of GSU1058 (sucC) were used as a probe. The DNA fragments used as a probe were prepared by PCR with primers 5′-TCTCTAGACAAGTTCCTGAAGTTCATTT-3′ and 5′-TCGGATCCAGTATTTCCTTTGCCTGGTACTC-3′ (fragment 1, Figure 5b), 5′-TCTCTAGACAAGTTCCTGAAGTTCATTT-3′ and 5′-TCGGATCCAACGCAATGCGAATGGTAA-3′ (fragment 2, Figure 5b), and 5′-TCTCTAGAAACTGTTTTATTAATACTA-3′ and 5′-TCGGATCCAGTATTTCCTTTGCCTGGTACTC-3′ (fragment 3, Figure 5b; XbaI and BamHI sites are underlined). The PCR products were digested with XbaI and BamHI and cloned into pET24b (Novagen). After confirmation of the sequence of the PCR products, the DNA fragment for the probe of the DNA-binding assay was prepared by digesting the plasmid with XbaI and BamHI and isolating the fragment by agarose gel electrophoresis. The isolated DNA fragment was labeled with (α-32P) dATP by Klenow fragment of DNA polymerase I (Feinberg and Vogelstein, 1983). DNA-binding assay was performed as described previously (Ueki and Inouye, 2005).
Footprint assay
DNA fragment containing the sucC promoter region was prepared with primers 5′-TCTCTAGAAGGCAAGATAGGACCA-3′ and 5′-TCGGATCCAGTATTTCCTTTGCCTGGTACTC-3′ (XbaI and BamHI sites are underlined) as described above with modification. The DNA fragment was prepared by digesting the plasmid with SacII and XhoI (XhoI is located in the plasmid vector). DNaseI footprint assays were conducted as described previously (Ueki and Inouye, 2005).
Results
Adaptation for growth on lactate associated with mutation in GSU0514
G. sulfurreducens was initially reported to be unable to grow on lactate with Fe(III) as an electron acceptor (Caccavo et al., 1994), but when cells grown with acetate as the electron donor and fumarate as the electron acceptor were inoculated (1% inoculum) into lactate-fumarate medium G. sulfurreducens grew slowly (doubling time 22 h) after a long lag period. This compared with a doubling time of 5 h in the same medium with acetate as the electron donor. However, with subsequent transfers of cultures in late logarithmic growth phase, five independent lines of serial transfers adapted for faster growth on lactate (Figure 1). The adaptive trajectories were not identical, but there was an overall trend of increased growth rate in all cultures. After 23 transfers (∼151 generations), the mean doubling time of the five replicates was 8.3 h and there was no longer a long, multiple day lag phase following inoculation into a fresh medium, as was observed with the first transfer on the lactate-fumarate medium.

Growth rates increased for all five (numbered) replicate strains selected for faster growth on lactate. The inverse of the growth rate is plotted versus the transfer number for each of the five replicate strains.
Sequencing the genomic DNA from individual clones from the 23rd transfer of the five replicates revealed a SNP in gene GSU0514 in all five replicates. No mutations in any other genes were detected. Three strains shared the same mutation and the two remaining strains each had a unique mutation (Figure 2a). In all five replicates the single-base-pair change resulted in a non-synonymous change in an amino acid (Figure 2b). The resulting amino acids that were changed in each of the five replicate strains were in conserved amino acids.

The single-base-pair mutations discovered in the five lactate-adapted replicates after 23 transfers in lactate media. (a) DNA sequence of Gsu0514 with non-synonymous SNPs found in the adapted strains highlighted in red, individual SNPs from the different replicate strains are all represented on the same sequence and labeled with arrows. (b) Amino acid sequence of selected regions from Gsu0514 containing mutations. The adapted strain with all of the replicate strains mutation represented in a single sequence is on top, followed by G. sulfurreducens wild-type sequence, and then aligned sequences from additional Geobacter species as well as Escherichia coli to show amino acid conservation across species for the mutated regions.
Single-base-pair mutation in Gsu0514 is sufficient for lactate adaptation
Genome resequencing may fail to detect transposition or other genome rearrangements (Zhong et al., 2004). Therefore, it was important to evaluate whether a single-base-pair mutation in GSU0514 could be sufficient for adaptation for rapid growth on lactate. The mutation found in replicate tube #1, designated strain ZMS123, and was chosen for further analysis because this strain grew the fastest of the five replicates whose genomes were sequenced. A strain in which the wild-type GSU0514 sequence was replaced with the sequence found in strain ZMS123 grew on lactate at nearly the same rate as strain ZMS123, with a doubling time of <10 h (Figure 3). However, because the single-base-pair change did not immediately result in the genetically engineered strain growing at the identical rate of ZMS123 on lactate, it should be noted that genome resequencing might fail to detect transposition or other genome rearrangements. However, these results demonstrate that a single-base-pair change in the genome can significantly enhance the organism's capacity for growth on lactate.

Growth curves on the NBLF media of G. sulfurreducens wild-type, adapted strain ZMS123 and the wild type with SNP from ZMS123 inserted on chromosome.
Identification of GSU0514 as a transcriptional regulator of the succinyl-CoA synthetase operon
Gsu0514 is annotated as a transcriptional regulator in the IclR family of transcriptional activators and repressors (Yan et al., 2007). The specific function of a novel IclR family protein cannot be predicted simply by sequence similarity (Krell et al., 2006; Molina-Henares et al., 2006).
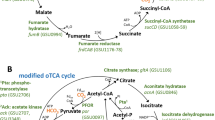
One potential difference in growth on lactate versus growth on acetate (Figure 4a) is the necessity for succinyl-CoA synthase for growth on lactate (Figure 4b). G. sulfurreducens oxidizes acetate via the tricarboxylic acid cycle (Galushko and Schink, 2000; Mahadevan et al., 2006). Acetate is activated to acetyl-CoA for introduction into the tricarboxylic acid cycle via succinyl-CoA:acetate-CoA-transferase (Galushko and Schink, 2000; Segura et al., 2008). Succinyl-CoA synthetase is not required for the conversion of succinyl CoA to succinate during growth on acetate and previous studies did not detect succinyl-CoA synthetase activity in acetate-grown cells (Galushko and Schink, 2000; Segura et al., 2008). However, when lactate is the electron donor, acetyl-CoA is produced directly from pyruvate and succinyl-CoA synthetase is required to recover the CoA while producing succinate and completing the tricarboxylic acid cycle (Figure 4b).

Metabolic pathways for G. sulfurreducens for (a) growth on acetate, without the necessity for succinyl-CoA synthetase and (b) growth on lactate with the succinyl-CoA synthetase being utilized. ACONT, aconitase; AKGO OOR, α-ketoglutarate oxidoreductase; ATO, acetyl CoA transferase; CS, citrate synthase; ICDHy, isocitrate dehydrogenase; FRDS, fumarate reductase/succinate dyhydrogenase; FUM, fumarase; LDH, lactate dehydrogenase; MDH, malate dehydrogenase; PDH pyruvate dehydrogenase; POR, pyruvate oxidoreductate; SUCOAS, succinyl-CoA synthetase.
During growth on lactate, transcript abundance of the genes for the two subunits of the succinyl-CoA transferase enzymes, GSU1058 (sucC) and GSU1059 (sucD), was significantly higher than in wild-type cells in each of the adaptively evolved strains, as well as in the strain in which the single-nucleotide polymorphism in GSU0514 of strain ZMS123 was genetically introduced (Figure 5). These results suggested that more succinyl-CoA synthetase is produced in the adapted strains, allowing the cell to completely oxidize the lactate.

Fold change in transcript abundance of the genes encoding the α (Gsu1058; SucC) and β (Gsu1059; SucD) subunits of the succinyl CoA synthetase enzyme in the five replicate strains at transfer 23 as well as the SNP knock-in train. All values are normalized to wild-type transcript abundance as determined with quantitative RT–PCR. Error bars illustrate the s.d. of the mean of triplicate determinations.
Footprint-binding assays demonstrated that the GSU0514 protein specifically bound to a putative promoter region for the sucC–sucD operon, (Figure 6a). The GSU0514-binding site, 5′-TATGGTATACATA-3′, is located downstream of putative transcription initiation sites and upstream of a translation initiation codon (Figure 6b). To further verify the identified GSU0514-binding site, DNA-binding assays were conducted with different regions in the promoter. GSU0514 bound only the DNA fragment containing the identified binding site (Figure 6c).

Binding of GSU0514 to the sucC–sucD promoter. (a) GSU0514-binding site. Footprint assays were carried out with GSU0514 and the upstream region of the sucC–sucD operon. DNA-binding reactions were conducted with GSU0514 (lane 1, 0 pmol; lane 2, 1.3 pmol and lane 3, 2.5 pmol) and probes (0.2 pmol). G, A, T and C represent sequence ladders. The region protected from or modified by DNaseI in the presence of GSU0514 is indicated by a vertical bar. (b) Promoter region of the sucC–sucD operon. The GSU0514-binding site identified by the footprint assay (a) is indicated by bold letters with a box. Two putative transcription initiation sites determined by primer extension assays (data not shown) are indicated by +1. Putative −35 and −10 promoter elements are indicated by −35 and −10, respectively. Putative ribosome-binding site is indicated by RBS. Translation initiation codon is indicated by Met. (c) DNA-binding assay. DNA fragments containing promoter regions of the sucC–sucD operon were tested for DNA-binding activity of GSU0514. The promoter regions in the DNA fragments (1∼3) used as a probe are shown below the diagram of the sucC–sucD operon. Arrows indicate putative transcription initiation sites. Black box represents the GSU0514-binding site identified by the footprint assay (a).
The location of the GSU0514-binding site in the sucC–sucD operon is consistent with the finding from the qRT-PCR analysis that mutations in GSU0514 enhanced expression of the sucC–sucD operon. Therefore, it is likely that GSU0514 is a transcriptional regulator that represses the transcription of the sucC–sucD operon by binding the sucC–sucD promoter and that mutations in GSU0514 negatively impact GSU0514 binding to the promoter. A search of the rest of the G. sulfurreducens genome failed to identify any additional sequences similar to the GSU0514-binding site in the sucC–sucD operon, suggesting that GSU0514 may directly regulate only the sucC–sucD operon.
Discussion
The results demonstrate that a single-base-pair change in a gene for a transcriptional regulator can markedly influence the metabolic capability of G. sulfurreducens. This finding offers insight into an adaptive strategy for microorganisms to enhance their utilization of substrates and suggests that artificial conditions imposed on subsurface microorganisms during bioremediation could readily result in an adaptive evolution response.
Laboratory evolution can be used as a discovery tool for identifying functions of previously unstudied genes that are linked to fitness increases during adaptation to selective laboratory environments. In the case of this study, a transcriptional regulator with previously unknown gene targets was identified, which when unable to bind to DNA, gives a fitness advantage to the cell in the form of increased fitness. This is because of the cells ability to more rapidly metabolize lactate, the sole electron donor and carbon source provided. Previous laboratory evolution studies have identified mutations in regulatory elements (Cooper et al., 2003; Crozat et al., 2005; Herring et al., 2006; Knight et al., 2006; D’Argenio et al., 2007; Giraud et al., 2008; Conrad et al., 2010), but these were all within global regulatory ‘hubs’ that subsequently affected gene expression on a large scale. In contrast, this study offers the finding of a mutated transcriptional regulator that appears to have a very narrow gene target, repression of two genes that encode for a single key enzyme. This further supports the observation that transcriptional regulatory networks are the first to change under selection (Shou et al., 2011), and at the same time presents a unique result in showing that a narrow-target regulator can also be involved in rapid evolution.
The possibility of a similar adaptive response occurring during bioremediation could have important implications for attempts to predict the response of the microbial community to bioremediation strategies. When genome-scale metabolic modeling is applied to bioremediation (Scheibe et al., 2009; Zhuang et al., 2010; Mahadevan et al., 2011) it may be possible to predict adaptive responses of the microbial species present (Edwards et al., 2001; Ibarra et al., 2002; Fong and Palsson, 2004; Teusink et al., 2009), and whether adaptive evolution is likely to yield a competitive advantage over other microorganisms that might already have the ability to use the added substrate. The increasing availability of strategies to deeply sequence genomic DNA in environmental samples may soon make it feasible to directly evaluate the possibility of adaptive evolution during bioremediation field experiments.
The finding that wild-type G. sulfurreducens only poorly metabolized lactate despite possessing the complete complement of genes required for this metabolism is one example of the difficulties faced when extrapolating metabolic function from genome sequence data alone. Information on the expression of the genes, or preferably, production of actual enzymes, is required before the actual metabolic capability of microorganisms can be inferred. This has important implications for the interpretation of metagenomic data from the subsurface and other environments.
References
Aklujkar M, Lovley DR . (2010). Interference with histidyl-tRNA synthetase by a CRISPR spacer sequence as a factor in the evolution of Pelobacter carbinolicus. BMC Evol Biol 10: 230.
Anderson RT, Vrionis HA, Ortiz-Bernad I, Resch CT, Long PE, Dayvault R et al. (2003). Stimulating the in situ activity of Geobacter species to remove uranium from the groundwater of a uranium-contaminated aquifer. Appl Environ Microbiol 69: 5884–5891.
Babu MM, Aravind L . (2006). Adaptive evolution by optimizing expression levels in different environments. Trends Microbiol 14: 11–14.
Babu MS, Veeranna, Raman P, Krishnan MG . (2007). Acute sarcoidosis--Heefordts-Waldenstrom syndrome. J Assoc Physicians India 55: 156–157.
Balch WE, Fox GE, Magrum LJ, Woese CR, Wolfe RS . (1979). Methanogens: reevaluation of a unique biological group. Microbiol Rev 43: 260–296.
Barrick JE, Yu DS, Yoon SH, Jeong H, Oh TK, Schneider D et al. (2009). Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature 461: 1243–1247.
Brockhurst MA, Colegrave N, Rozen DE . (2010). Next-generation sequencing as a tool to study microbial evolution. Mol Ecol 20: 972–980.
Caccavo Jr F, Lonergan DJ, Lovley DR, Davis M, Stolz JF, McInerney MJ . (1994). Geobacter sulfurreducens sp. nov., a hydrogen- and acetate-oxidizing dissimilatory metal-reducing microorganism. Appl Environ Microbiol 60: 3752–3759.
Chang YJ, Long PE, Geyer R, Peacock AD, Resch CT, Sublette K et al. (2005). Microbial incorporation of 13C-labeled acetate at the field scale: detection of microbes responsible for reduction of U(VI). Environ Sci Technol 39: 9039–9048.
Conrad TM, Frazier M, Joyce AR, Cho BK, Knight EM, Lewis NE et al. (2010). RNA polymerase mutants found through adaptive evolution reprogram Escherichia coli for optimal growth in minimal media. Proc Natl Acad Sci USA 107: 20500–20505.
Conrad TM, Joyce AR, Applebee MK, Barrett CL, Xie B, Gao Y et al. (2009). Whole-genome resequencing of Escherichia coli K-12 MG1655 undergoing short-term laboratory evolution in lactate minimal media reveals flexible selection of adaptive mutations. Genome Biol 10: R118.
Cooper TF, Rozen DE, Lenski RE . (2003). Parallel changes in gene expression after 20,000 generations of evolution in Escherichia coli. Proc Natl Acad Sci USA 100: 1072–1077.
Coppi MV, Leang C, Sandler SJ, Lovley DR . (2001). Development of a genetic system for Geobacter sulfurreducens. Appl Environ Microbiol 67: 3180–3187.
Crozat E, Philippe N, Lenski RE, Geiselmann J, Schneider D . (2005). Long-term experimental evolution in Escherichia coli. XII. DNA topology as a key target of selection. Genetics 169: 523–532.
D’Argenio DA, Wu M, Hoffman LR, Kulasekara HD, Deziel E, Smith EE et al. (2007). Growth phenotypes of Pseudomonas aeruginosa lasR mutants adapted to the airways of cystic fibrosis patients. Mol Microbiol 64: 512–533.
Dekel E, Alon U . (2005). Optimality and evolutionary tuning of the expression level of a protein. Nature 436: 588–592.
Edwards JS, Ibarra RU, Palsson BO . (2001). In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat Biotechnol 19: 125–130.
Feinberg AP, Vogelstein B . (1983). A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem 132: 6–13.
Ferea TL, Botstein D, Brown PO, Rosenzweig RF . (1999). Systematic changes in gene expression patterns following adaptive evolution in yeast. Proc Natl Acad Sci USA 96: 9721–9726.
Finneran K, Anderson R, Nevin K, Lovley D . (2002). Potential for bioremediation of uranium-contaminated aquifers with microbial U(VI) reduction. Soil Sediment Contam 11: 339–357.
Fong SS, Burgard AP, Herring CD, Knight EM, Blattner FR, Maranas CD et al. (2005a). In silico design and adaptive evolution of Escherichia coli for production of lactic acid. Biotechnol Bioeng 91: 643–648.
Fong SS, Joyce AR, Palsson BO . (2005b). Parallel adaptive evolution cultures of Escherichia coli lead to convergent growth phenotypes with different gene expression states. Genome Res 15: 1365–1372.
Fong SS, Palsson BO . (2004). Metabolic gene-deletion strains of Escherichia coli evolve to computationally predicted growth phenotypes. Nat Genet 36: 1056–1058.
Galushko AS, Schink B . (2000). Oxidation of acetate through reactions of the citric acid cycle by Geobacter sulfurreducens in pure culture and in syntrophic coculture. Arch Microbiol 174: 314–321.
Giraud A, Arous S, De Paepe M, Gaboriau-Routhiau V, Bambou JC, Rakotobe S et al. (2008). Dissecting the genetic components of adaptation of Escherichia coli to the mouse gut. PLoS Genet 4: e2.
Hazen TC, Tabak HH . (2005). Developments in bioremediation of soils and sediments polluted with metals and radionuclides: 2. Field research on bioremediation of metals and radionuclides. Rev Environ Sci Biotech 4: 157–183.
Herring CD, Raghunathan A, Honisch C, Patel T, Applebee MK, Joyce AR et al. (2006). Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale. Nat Genet 38: 1406–1412.
Holmes DE, O’Neil RA, Chavan MA, N’Guessan LA, Vrionis HA, Perpetua LA et al. (2009). Transcriptome of Geobacter uraniireducens growing in uranium-contaminated subsurface sediments. ISME J 3: 216–230.
Ibarra RU, Edwards JS, Palsson BO . (2002). Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature 420: 186–189.
Knight CG, Zitzmann N, Prabhakar S, Antrobus R, Dwek R, Hebestreit H et al. (2006). Unraveling adaptive evolution: how a single point mutation affects the protein coregulation network. Nat Genet 38: 1015–1022.
Kovach ME, Phillips RW, Elzer PH, Roop II RM, Peterson KM . (1994). pBBR1MCS: a broad-host-range cloning vector. Biotechniques 16: 800–802.
Krell T, Molina-Henares AJ, Ramos JL . (2006). The IclR family of transcriptional activators and repressors can be defined by a single profile. Protein Sci 15: 1207–1213.
Lee DH, Palsson BO . (2010). Adaptive evolution of Escherichia coli K-12 MG1655 during growth on a Nonnative carbon source, L-1,2-propanediol. Appl Environ Microbiol 76: 4158–4168.
Lovley DR . (2003). Cleaning up with genomics: applying molecular biology to bioremediation. Nat Rev Microbiol 1: 35–44.
Lovley DR, Chapelle FH . (1995). Deep subsurface microbial processes. Revi Geophys 33: 365–381.
Mahadevan R, Bond DR, Butler JE, Esteve-Nunez A, Coppi MV, Palsson BO et al. (2006). Characterization of metabolism in the Fe(III)-reducing organism Geobacter sulfurreducens by constraint-based modeling. Appl Environ Microbiol 72: 1558–1568.
Mahadevan R, Palsson BO, Lovley DR . (2011). In situ to in silico and back: elucidating the physiology and ecology of Geobacter spp. using genome-scale modelling. Nat Rev Microbiol 9: 39–50.
Marx CJ, Lidstrom ME . (2002). Broad-host-range cre-lox system for antibiotic marker recycling in gram-negative bacteria. Biotechniques 33: 1062–1067.
Methe BA, Nelson KE, Eisen JA, Paulsen IT, Nelson W, Heidelberg JF et al. (2003). Genome of Geobacter sulfurreducens: metal reduction in subsurface environments. Science 302: 1967–1969.
Molina-Henares AJ, Krell T, Eugenia Guazzaroni M, Segura A, Ramos JL . (2006). Members of the IclR family of bacterial transcriptional regulators function as activators and/or repressors. FEMS Microbiol Rev 30: 157–186.
N’Guessan AL, Vrionis HA, Resch CT, Long PE, Lovley DR . (2008). Sustained removal of uranium from contaminated groundwater following stimulation of dissimilatory metal reduction. Environ Sci Technol 42: 2999–3004.
Pelosi L, Kuhn L, Guetta D, Garin J, Geiselmann J, Lenski RE et al. (2006). Parallel changes in global protein profiles during long-term experimental evolution in Escherichia coli. Genetics 173: 1851–1869.
Philippe N, Crozat E, Lenski RE, Schneider D . (2007). Evolution of global regulatory networks during a long-term experiment with Escherichia coli. Bioessays 29: 846–860.
Reguera G, McCarthy KD, Mehta T, Nicoll JS, Tuominen MT, Lovley DR . (2005). Extracellular electron transfer via microbial nanowires. Nature 435: 1098–1101.
Reguera G, Nevin KP, Nicoll JS, Covalla SF, Woodard TL, Lovley DR . (2006). Biofilm and nanowire production leads to increased current in Geobacter sulfurreducens fuel cells. Appl Environ Microbiol 72: 7345–7348.
Scheibe TD, Mahadevan R, Fang Y, Garg S, Long PE, Lovley DR . (2009). Coupling a genome-scale metabolic model with a reactive transport model to describe in situ uranium bioremediation. Microb Biotechnol 2: 274–286.
Segura D, Mahadevan R, Juarez K, Lovley DR . (2008). Computational and experimental analysis of redundancy in the central metabolism of Geobacter sulfurreducens. PLoS Comput Biol 4: e36.
Shaver AC, Dombrowski PG, Sweeney JY, Treis T, Zappala RM, Sniegowski PD . (2002). Fitness evolution and the rise of mutator alleles in experimental Escherichia coli populations. Genetics 162: 557–566.
Shou C, Bhardwaj N, Lam HY, Yan KK, Kim PM, Snyder M et al. (2011). Measuring the evolutionary rewiring of biological networks. PLoS Comput Biol 7: e1001050.
Teusink B, Wiersma A, Jacobs L, Notebaart RA, Smid EJ . (2009). Understanding the adaptive growth strategy of Lactobacillus plantarum by in silico optimisation. PLoS Comput Biol 5: e1000410.
Tremblay PL, Summers ZM, Glaven RH, Nevin KP, Zengler K, Barrett CL et al. (2011). A c-type cytochrome and a transcriptional regulator responsible for enhanced extracellular electron transfer in Geobacter sulfurreducens revealed by adaptive evolution. Environ Microbiol 13: 13–23.
Treves DS, Manning S, Adams J . (1998). Repeated evolution of an acetate-crossfeeding polymorphism in long-term populations of Escherichia coli. Mol Biol Evol 15: 789–797.
Ueki T, Inouye S . (2002). Transcriptional activation of a heat-shock gene, lonD, of Myxococcus xanthus by a two component histidine-aspartate phosphorelay system. J Biol Chem 277: 6170–6177.
Ueki T, Inouye S . (2005). Identification of a gene involved in polysaccharide export as a transcription target of FruA, an essential factor for Myxococcus xanthus development. J Biol Chem 280: 32279–32284.
Wick LM, Weilenmann H, Egli T . (2002). The apparent clock-like evolution of Escherichia coli in glucose-limited chemostats is reproducible at large but not at small population sizes and can be explained with Monod kinetics. Microbiology 148: 2889–2902.
Yan B, Lovley DR, Krushkal J . (2007). Genome-wide similarity search for transcription factors and their binding sites in a metal-reducing prokaryote Geobacter sulfurreducens. Biosystems 90: 421–441.
Zhong S, Khodursky A, Dykhuizen DE, Dean AM . (2004). Evolutionary genomics of ecological specialization. Proc Natl Acad Sci USA 101: 11719–11724.
Zhuang K, Izallalen M, Mouser P, Richter H, Risso C, Mahadevan R et al. (2010). Genome-scale dynamic modeling of the competition between Rhodoferax and Geobacter in anoxic subsurface environments. ISME J 5: 305–316.
Acknowledgements
We thank P Brown, C Barrett, K Zengler and Y Qiu for sequencing assistance. This work was supported by the Office of Science (BER), the US Department of Energy, grants DE-FC02-02ER63446 and DE-SC0004080.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Summers, Z., Ueki, T., Ismail, W. et al. Laboratory evolution of Geobacter sulfurreducens for enhanced growth on lactate via a single-base-pair substitution in a transcriptional regulator. ISME J 6, 975–983 (2012). https://doi.org/10.1038/ismej.2011.166
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2011.166
Keywords
This article is cited by
-
Set-up of a pharmaceutical cell bank of Magnetospirillum gryphiswaldense MSR1 magnetotactic bacteria producing highly pure magnetosomes
Microbial Cell Factories (2024)
-
Moving towards the enhancement of extracellular electron transfer in electrogens
World Journal of Microbiology and Biotechnology (2023)
-
Adapted laboratory evolution of Thermotoga sp. strain RQ7 under carbon starvation
BMC Research Notes (2022)
-
The hidden chemolithoautotrophic metabolism of Geobacter sulfurreducens uncovered by adaptation to formate
The ISME Journal (2020)
-
Recent Advances in Microbial Cell Growth Regulation Strategies for Metabolic Engineering
Biotechnology and Bioprocess Engineering (2020)
