
Abstract
Magnetotactic bacteria (MTB), which synthesize intracellular ferromagnetic magnetite and/or greigite magnetosomes, have significant roles in global iron cycling in aquatic systems, as well as sedimentary magnetism. The occurrence of MTB has been reported in aquatic environments from freshwater to marine ecosystems; however, the distribution of MTB across heterogeneous habitats remains unclear. Here we examined the MTB communities from diverse habitats across northern and southern China, using comprehensive transmission electron microscopy and comparison of 16S rRNA gene analyses. A total of 334 16S rRNA gene sequences were analyzed, representing the most comprehensive analysis on the diversity and distribution of MTB to date. The majority (95%) of sequences belong to the Alphaproteobacteria, whereas a population of giant magnetotactic rod is affiliated with the Nitrospirae phylum. By a statistical comparison of these sequence data and publicly available MTB sequences, we infer for the first time that the composition of MTB communities represents a biogeographic distribution across globally heterogeneous environments, which is influenced by salinity.
Similar content being viewed by others

Main
Magnetotactic bacteria (MTB) are a diverse group of microorganisms, which are ubiquitous in aquatic environments from freshwater to marine ecosystems (Bazylinski and Frankel, 2004). These bacteria can mineralize intracellular nanosized iron oxides (Fe3O4) and/or sulfides (Fe3S4) named magnetosomes (Jogler and Schüler, 2009). Based on the analysis of 16S rRNA genes, all known MTB are affiliated within Proteobacteria and Nitrospirae phyla (Amann et al., 2006). Considering their high abundance (104–106 cells perml) near the oxic–anoxic transition zone and high intracellular iron content (10−13 to 10−15 g per cell), MTB have been attributed as an important role in iron cycling in aquatic systems (Faivre and Schüler, 2008). Moreover, fossil magnetosomes preserved in sediments may significantly contribute to the bulk magnetization of sediments and serve as potential archives of paleoenvironmental information (Kopp and Kirschvink, 2008; Lin and Pan, 2009). However, due to the lack of comprehensive study on the distribution of MTB across different ecosystems thus far, we know little about the biogeography of MTB communities.
In the present study, we analyzed the MTB communities from different ecosystems across northern and southern China, including four freshwater lakes, two mangrove swamps, two estuaries and one intertidal zone, with a salinity gradient ranging from 0.18 to 28.6 parts per thousand (p.p.t.) and spanning latitudes of 19°N–40°N. MTB were magnetically enriched solely using the ‘MTB trap’ method, which simultaneously collected both north- and south-seeking MTB (Jogler et al., 2009). After magnetic collection, the enriched north- and south-seeking MTB from same sample were pooled. The diversity of MTB communities in each site was examined by transmission electron microscopy and comparison of 16S rRNA genes. Detailed descriptions of sampling sites and experimental procedures are given as Supplementary Information. MTB communities from different sites in this study, along with publicly available data, were further compared using statistically phylogenetic methods.
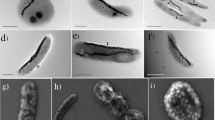
Live MTB were enriched from all nine sampling sites. Only coccoid MTB were observed in the saline samples from the mangrove swamps, estuaries and intertidal zone (Figures 1a–c). However, great morphological variability was observed in the MTB enrichment from the freshwater lakes, including cocci (Figures 1d–f), vibrios (Figures 1g and h) and a giant rod-shaped bacterium (Figure 1i); this rod was morphologically similar to ‘Candidatus Magnetobacterium bavaricum’, found in Lake Chiemsee (Spring et al., 1993; Jogler et al., 2010, 2011), and ‘Ca. Magnetobacterium bavaricum’-like MTB found in Lake Miyun previously (Lin et al., 2011).

Representative transmission electron micrographs of various MTB from nine locations across northern and southern China. (a–c) are MTB from saline environments, including the mangrove swamps, estuaries and intertidal zone. (d–i) are MTB from freshwater lakes. Bars=500 nm.
The 16S rRNA genes were directly amplified from magnetically enriched MTB, using the bacterial universal primers 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′) as previously described (Lin et al., 2008). Randomly selected clones were sequenced using a 27F primer (Beijing Genomics Institute, Beijing, China). After removing sequences of insufficient length (<400 bp) and low quality, nearly 400 sequences were retrieved in this study. The presence of potential chimeras was checked using the Greengenes chimera-check tool (Huber et al., 2004). Twenty-six sequences, which were most similar to non-MTB bacteria, but unrelated to known MTB (<80% sequence identity), were attributed to contamination by non-magnetotactic organisms (Supplementary Information and Supplementary Table 3), and were thus removed from the analyses. In this way, a total of 334 high quality 16S rRNA genes were retrieved, which covered V1 to V3 hypervariable regions (450–500 bp) and sufficed for accurate microbial diversity analysis (Liu et al., 2007).
These sequences were composed of 43 operational taxonomic units (OTUs) defined as 98% similarity level, representing only 3–11 OTUs per location (Figure 2a and Supplementary Table 1). Although the potential limit of the magnetic enrichment approach (Lin et al., 2008) and/or the strict MTB sequences assignment applied in this study may restrict the assessment of MTB diversity, our results were similar to the diversity level reported elsewhere (Simmons et al., 2004; Flies et al., 2005). Rarefaction curves for all nine libraries nearly reached an asymptote, indicating that we have captured the major extent of MTB diversity (Figure 2a). Therefore, our results likely reflect the true situation that the sequence diversity of MTB communities in nature, compared with the whole bacterial community (Whitman et al., 1998), is not very high. Out of 334 sequences, 318 sequences (42 OTUs) were affiliated within the Alphaproteobacteria, whereas the residual 16 sequences (1 OTU) detected from freshwater sediments belonged to the Nitrospirae phylum and were 97.9% similar to ‘Ca. Magnetobacterium bavaricum’ (Figure 2b). The phylogenetic structure of all OTUs retrieved here suggested a biogeographic distribution of MTB communities. For example, out of 43 OTUs, 31 OTUs were endemic, whereas none were cosmopolitan (Figure 2b). In addition, there was no overlap in OTUs between freshwater and saline environments.

(a) Rarefaction curves for individual libraries of the highest, medium and lowest number of OTUs. (b) Neighbor-joining phylogenetic tree and relative abundances of 43 OTUs (98% sequence similarity) retrieved from the nine locations across northern and southern China. Bootstrap values were indicated at nodes. (c) Principal coordinates analysis of unweighted UniFrac distance matrix showing the overall phylogenetic similarity of the MTB communities examined in this study. In this analysis, previously reported MTB communities from Itaipu Lagoon in Rio de Janeiro (Brazil), Jiaozhou Bay in Shandong Province (China) and Lake Chiemsee near Munich (Germany) were included to compare their phylogenetic relationships with the MTB communities retrieved in this study. The first principal axis (PC1) is dominated by salinity, indicating the key effect of salinity on the distribution of MTB communities.
To understand the global biogeographic pattern of MTB communities, we compared each community of this study and publicly available MTB sequence sets from Itaipu lagoon (saline) in Brazil, Jiaozhou Bay (saline) in China and Lake Chiemsee (freshwater) in Germany, with a matrix of UniFrac distances (Hamady et al., 2010) through principal coordinates analysis (Supplementary Table 2). Of particular interest, all MTB communities were grouped by salinity rather than geographic distances or continents (Figure 2c). Among them, MTB communities from freshwater environments, even those from Lake Chiemsee in Germany, which is geographically distant from China, clustered together along principal coordinate 1. On the other hand, MTB communities from the saline sediments, including Itaipu Lagoon in the Southern Hemisphere, were more similar to each other than to their freshwater counterparts. The salinity-dependent distribution of MTB was further confirmed by Spearman rank correlations analysis, which demonstrated that the salinity was significantly (Spearman’s ρ=0.619, P=0.003) correlated with the degree of community distance across the nine Chinese sampling sites. These community distances were not significantly related to other factors that were measured, such as pH, oxygen concentration and temperature (P>0.05). Despite the small number of samplings, to our knowledge, this is the most comprehensive study on the diversity and distribution of dominant MTB clades across a large spatial scale to date, which, for the first time, shows that salinity has a strong influence on the biogeography of MTB. Our results support the view that bacteria, like plants and animals, are not globally homogeneous, but represent biogeographies (Martiny et al., 2006), which are primarily influenced by salinity (Lozupone and Knight, 2007).
The correlation between salinity and MTB communities observed here raises the question: why can salinity influence the distribution of MTB? One hypothesis is that different salinities (and their related osmotic pressure) can affect the energetic cost and metabolic pathways of microorganisms (Oren, 2001), including MTB communities. In addition to salinity, it is possible that other geochemical factors that co-vary with salinity, or even local competitors and predators, may affect the distribution of MTB. The geographic distance among sites does not seem to significantly influence MTB community composition according to our results. High dispersal capacity, making geographic distance irrelevant to the incidence of MTB, is a possible explanation. This result represents the famous microbiological tenet ‘everything is everywhere, but, the environment selects’, the so-called Baas-Becking hypothesis (de Wit and Bouvier, 2006). That is, MTB are probably widely dispersed over great distances or may be robust over long-distance transport, and environmental heterogeneity (like salinity) determines their ability to thrive within specific environments. Nevertheless, the true causes for salinity-dependent distribution of MTB communities across different continents needs to be further studied. Knowledge of the biogeographic distribution of MTB will help to better understand the global iron dynamics in aquatic environments, and perhaps can also be applied towards the reconstruction of the paleoenvironment, based on the fossil magnetosomes.
Nucleotide sequence accession numbers
The sequence data has been submitted to the DDBJ/EMBL/GenBank databases under accession numbers HQ437323-HQ437656.
Accession codes
References
Amann R, Peplies J, Schüler D . (2006). Diversity and taxonomy of magnetotactic bacteria. In: Schüler D (ed). Magnetoreception and Magnetosomes in Bacteria. Springer: Berlin, pp 25–36.
Bazylinski DA, Frankel RB . (2004). Magnetosome formation in prokaryotes. Nat Rev Microbiol 2: 217–230.
de Wit R, Bouvier T . (2006). ‘Everything is everywhere, but, the environment selects’; what did Baas Becking and Beijerinck really say? Environ Microbiol 8: 755–758.
Faivre D, Schüler D . (2008). Magnetotactic bacteria and magnetosomes. Chem Rev 108: 4875–4898.
Flies CB, Peplies J, Schüler D . (2005). Combined approach for characterization of uncultivated magnetotactic bacteria from various aquatic environments. Appl Environ Microbiol 71: 2723–2731.
Hamady M, Lozupone CA, Knight R . (2010). Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J 4: 17–27.
Huber T, Faulkner G, Hugenholtz P . (2004). Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20: 2317–2319.
Jogler C, Lin W, Meyerdierks A, Kube M, Katzmann E, Flies C et al. (2009). Towards cloning the magnetotactic metagenome: identification of magnetosome island gene clusters in uncultivated magnetotactic bacteria from different aquatic sediments. Appl Environ Microbiol 75: 3972–3979.
Jogler C, Niebler M, Lin W, Kube M, Wanner G, Kolinko S et al. (2010). Cultivation-independent characterization of ‘Candidatus Magnetobacterium bavaricum’ via ultrastructural, geochemical, ecological and metagenomic methods. Environ Microbiol 12: 2466–2478.
Jogler C, Schüler D . (2009). Genomics, genetics, and cell biology of magnetosome formation. Annu Rev Microbiol 63: 501–521.
Jogler C, Wanner G, Kolinko S, Niebler M, Amann R, Petersen N et al. (2011). Conservation of proteobacterial magnetosome genes and structures in an uncultivated member of the deep-branching Nitrospira phylum. Proc Natl Acad Sci USA 108: 1134–1139.
Kopp RE, Kirschvink JL . (2008). The identification and biogeochemical interpretation of fossil magnetotactic bacteria. Earth-Sci Rev 86: 42–61.
Lin W, Jogler C, Schüler D, Pan Y . (2011). Metagenomic analysis reveals unexpected subgenomic diversity of magnetotactic bacteria within the Phylum Nitrospirae. Appl Environ Microbiol 77: 323–326.
Lin W, Pan Y . (2009). Uncultivated magnetotactic cocci from Yuandadu park in Beijing, China. Appl Environ Microbiol 75: 4046–4052.
Lin W, Tian L, Li J, Pan Y . (2008). Does capillary racetrack-based enrichment reflect the diversity of uncultivated magnetotactic cocci in environmental samples? FEMS Microbiol Lett 279: 202–206.
Liu ZZ, Lozupone CA, Hamady M, Bushman FD, Knight R . (2007). Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res 35: e120.
Lozupone CA, Knight R . (2007). Global patterns in bacterial diversity. Proc Natl Acad Sci USA 104: 11436–11440.
Martiny JBH, Bohannan BJM, Brown JH, Colwell RK, Fuhrman JA, Green JL et al. (2006). Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol 4: 102–112.
Oren A . (2001). The bioenergetic basis for the decrease in metabolic diversity at increasing salt concentrations: implications for the functioning of salt lake ecosystems. Hydrobiologia 466: 61–72.
Simmons SL, Sievert SM, Frankel RB, Bazylinski DA, Edwards KJ . (2004). Spatiotemporal distribution of marine magnetotactic bacteria in a seasonally stratified coastal salt pond. Appl Environ Microbiol 70: 6230–6239.
Spring S, Amann R, Ludwig W, Schleifer KH, van Gemerden H, Petersen N . (1993). Dominating role of an unusual magnetotactic bacterium in the microaerobic zone of a freshwater sediment. Appl Environ Microbiol 59: 2397–2403.
Whitman WB, Coleman DC, Wiebe WJ . (1998). Prokaryotes: the unseen majority. Proc Natl Acad Sci USA 95: 6578–6583.
Acknowledgements
We would like to thank Jing Zhang, Zhuoyi Zhu, Ruifeng Zhang and Hongyan Bao in the East China Normal University, for help in field sampling. We are grateful to the anonymous reviewers for their very useful comments on an earlier version of the manuscript. We also thank Dirk Schüler for valuable comments, and Andrea Cheung for improving the English grammar on the revised version of the manuscript. This work was funded by the CAS/SAFEA International Partnership Program for Creative Research Teams (KZCX2-YW-T10) and NSFC grant (40821091).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Rights and permissions
About this article
Cite this article
Lin, W., Wang, Y., Li, B. et al. A biogeographic distribution of magnetotactic bacteria influenced by salinity. ISME J 6, 475–479 (2012). https://doi.org/10.1038/ismej.2011.112
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2011.112
Keywords
This article is cited by
-
Renaissance for magnetotactic bacteria in astrobiology
The ISME Journal (2023)
-
Morphological and phylogenetic diversity of magnetotactic bacteria and multicellular magnetotactic prokaryotes from a mangrove ecosystem in the Sanya River, South China
Journal of Oceanology and Limnology (2021)
-
Characterization and diversity of magnetotactic bacteria from sediments of Caroline Seamount in the Western Pacific Ocean
Journal of Oceanology and Limnology (2021)
-
Diversity of Uncultured Magnetospirillum sp. from the Sediments of South Kerala Sedimentary Basin, India
Current Microbiology (2020)
-
Distribution and diversity of magnetotactic bacteria in sediments of the Yellow Sea continental shelf
Journal of Soils and Sediments (2018)
