
Abstract
Methane emission from peatlands contributes substantially to global warming but is significantly reduced by sulfate reduction, which is fuelled by globally increasing aerial sulfur pollution. However, the biology behind sulfate reduction in terrestrial ecosystems is not well understood and the key players for this process as well as their abundance remained unidentified. Comparative 16S rRNA gene stable isotope probing (SIP) in the presence and absence of sulfate indicated that a Desulfosporosinus species, which constitutes only 0.006% of the total microbial community 16S rRNA genes, is an important sulfate reducer in a long-term experimental peatland field site. Parallel SIP using dsrAB (encoding subunit A and B of the dissimilatory (bi)sulfite reductase) identified no additional sulfate reducers under the conditions tested. For the identified Desulfosporosinus species a high cell-specific sulfate reduction rate of up to 341 fmol SO42− cell−1 day−1 was estimated. Thus, the small Desulfosporosinus population has the potential to reduce sulfate in situ at a rate of 4.0–36.8 nmol (g soil w. wt.)−1 day−1, sufficient to account for a considerable part of sulfate reduction in the peat soil. Modeling of sulfate diffusion to such highly active cells identified no limitation in sulfate supply even at bulk concentrations as low as 10 μM. Collectively, these data show that the identified Desulfosporosinus species, despite being a member of the ‘rare biosphere’, contributes to an important biogeochemical process that diverts the carbon flow in peatlands from methane to CO2 and, thus, alters their contribution to global warming.
Similar content being viewed by others


Introduction
Peatlands harbor up to one-third of the world pool of soil carbon (Limpens et al., 2008) and are estimated to be responsible for 10–20% of the global emission of the greenhouse gas methane (Houweling et al., 1999; Wuebbles and Hayhoe, 2002). Although regarded as a primarily methanogenic environment, dissimilatory sulfate reduction contributes up to 36% of carbon mineralization in these ecosystems, depending on sulfur deposition by rain or groundwater (Vile et al., 2003; Blodau et al., 2007; Deppe et al., 2009). Sulfate concentrations in peatlands are generally low, being in the range of 10–300 μM (Blodau et al., 2007; Schmalenberger et al., 2007; Deppe et al., 2009). However, the turnover time of the standing sulfate pool can be less than a day (Blodau et al., 2007; Knorr and Blodau, 2009), indicating a rapid recycling mechanism. Recycling of sulfate can proceed by the aerobic oxidation of sulfide, for example, in regions in which oxygen penetration and anoxic microniches overlap (in the zone above water saturation) (Knorr and Blodau, 2009; Knorr et al., 2009), in the rhizosphere of aerenchym-containing plants (Wind and Conrad, 1997) or during drying–rewetting events (Knorr and Blodau, 2009; Knorr et al., 2009; Reiche et al., 2009). In addition, experimental evidence is gathering for a rapid anoxic recycling mediated by oxidation of sulfide mainly with quinone moieties of the large pool of humic matter in peatlands (Jørgensen, 1990a, 1990b; Heitmann and Blodau, 2006; Blodau et al., 2007; Heitmann et al., 2007) or by electric currents spanning from the anoxic to the oxic zone (Nielsen et al., 2010).
This sulfate recycling is important, as sulfate reducers compete for substrates with microorganisms involved in the methanogenic degradation pathway, resulting in a considerable diversion of the carbon flow in peatlands from methane to CO2 (Gauci et al., 2004). In the near future, this effect will become even more pronounced because population growth and increasing energy consumption in Asia as well as exploitation and combustion of oil sands are predicted to increase global sulfur deposition on peatlands by acid rain (Gauci et al., 2004; Limpens et al., 2008). Furthermore, proposed geo-engineering solutions to counteract global warming through SO2 deposition into the stratosphere (Rasch et al., 2008) would additionally increase terrestrial sulfur deposition and soil acidification. Despite the de-acidification function of terrestrial sulfate reducers (Alewell et al., 2008) and their predicted suppression of global methane emission from peatlands by up to 15% (Gauci et al., 2004), we know very little about these microorganisms. To identify active sulfate reducers, we studied a peatland that is part of a long-term experimental field site in the German-Czech border region (Supplementary Figure 1) and was exposed to extensive acid rain and sulfur deposition during the time of intensive soft coal burning in Eastern Europe (Moldan and Schnoor, 1992; Berge et al., 1999). In anoxic peat soil incubations of this particular site, sulfate reduction can cause a decrease in methanogenesis by 68–78% (Loy et al., 2004). In this study, we present cumulative evidence that a low G+C, Gram-positive member of the ‘rare biosphere’ is an important sulfate reducer in this minerotrophic peatland.
Materials and methods
Sampling site
The minerotrophic fen Schlöppnerbrunnen II (50°08′38′′N, 11°51′41′′E), which was used as a model habitat for peatlands in this study, is situated in the Fichtelgebirge Mountains in northeastern Bavaria, Germany (Supplementary Figure 1). The site has been studied extensively over the past decades (see for example, Loy et al., 2004; Matzner, 2004; Paul et al., 2006; Schmalenberger et al., 2007). In brief, the soil pH is typically at pH 4–5 (Loy et al., 2004; Küsel et al., 2008; Reiche et al., 2009) and sulfate concentrations vary from 20 to 240 μM (Loy et al., 2004; Schmalenberger et al., 2007). Standing pools of lactate, acetate and formate are generally in the lower μM range (up to 100, 100 and 190 μM, respectively) but can reach peak concentrations of 3.2 mM (acetate) and 1.4 mM (formate). Propionate is only occasionally detected but can reach peak concentrations of 1.6 mM (Schmalenberger et al., 2007; Küsel et al., 2008). Sampling is detailed in Supplementary Methods.
Preincubation and stable isotope labeling
Incubations were set up to mimic the conditions in the peatland as closely as possible with respect to in situ concentrations of sulfate and substrates. For this purpose, 30 g of soil from the 10- to 20-cm depth fraction was gassed in a 125-ml serum bottle with N2 and mixed under the same N2 stream with 60 ml of filter-sterilized (0.2 μM) anoxic fen water. Subsequently, serum bottles were sealed with butyl rubber septa and incubated without agitation at 14 °C in the dark. The soil slurries had a pH of 4. For substrate turnover determination and stable isotope labeling, an unlabeled or fully 13C-labeled substrate mixture was added weekly to the mesocosms. The mixture consisted of lactate, acetate, formate and propionate (end concentration 50–200 μM each). In addition, sulfate was added weekly to an end concentration of 100–200 μM. Soil slurries with substrate but without sulfate addition served as controls. Upon substrate addition, soil slurries were briefly shaken to ensure complete mixing. The turnover of added substrates and sulfate was measured by ion chromatography as detailed in Supplementary Methods.
Stable isotope probing (SIP)
For SIP analyses, total nucleic acids were extracted from frozen samples (–80 °C) by grinding in liquid nitrogen and following thereafter the procedure described by Lüders et al. (2004). Minor modifications included a humic acid precipitation step with 7.5 M Na-acetate as described by Bodrossy et al. (2006). DNA was separated from RNA using the AllPrep DNA/RNA Mini Kit (Qiagen, Hilden, Germany) and quantified using PicoGreen staining according to the manufacturer's protocol (Invitrogen, Lofer, Austria). Density gradient centrifugation was performed as described by Neufeld et al. (2007). Gradients were fractionated into 20 equal fractions (ca. 250 μl); 50-μl aliquots of each fraction were used for density determination using a refractometer (AR 200; Reichert Analytical Instruments, Depew, NY, USA). DNA was extracted from fractions as described previously by Lüders et al. (2004). Terminal restriction fragment length polymorphism (T-RFLP) analysis as well as amplification, cloning and phylogenetic analysis of 16S rRNA genes and dsrAB (encoding subunit A and B of the dissimilatory (bi)sulfite reductase) are detailed in Supplementary Methods.
Quantitative real-time PCR (qPCR) analysis
For qPCR analysis of pristine non-incubated soil samples, DNA was extracted from 250 mg of peat soil (wet weight) using the Power Soil DNA Kit (MoBio Laboratories, Solana Beach, CA, USA). Desulfosporosinus-targeted and total Bacteria/Archaea-targeted qPCRs of 16S rRNA genes were performed using the primer pairs DSP603F (5′-TGTGAAAGATCAGGGCTCA-3′)/DSP821R (5′-CCTCTACACCTAGCACTC-3′) (constructed based on clone libraries of this study and the Arb SILVA 96 database (Pruesse et al., 2007)) and modified 1389F (5′-TGTACACACCGCCCGT-3′)/1492R (5′-GGYTACCTTGTTACGACTT-3′) (Loy et al., 2002), respectively. Primer 1389F has a weak mismatch to archaeal 16S rRNA genes at the third position from the 5′-end (T vs C) but is not regarded to be discriminative against Archaea. Reactions were performed in triplicates using the Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen), fluorescein (10 nM), bovine serum albumine (5 μg μl−1), 5 ng of template DNA and the following annealing temperatures: 64 °C for Desulfosporosinus and 52 °C for Bacteria/Archaea. Further details are given in Supplementary Methods.
Nucleic acid sequences
16S rRNA gene and dsrAB sequences obtained from the ‘heaviest’ PCR-amplifiable SIP fractions have been deposited at GenBank under accession numbers GU270657–GU270832 and GU371932–GU372082, respectively. The dsrAB sequence of Desulfosporosinus strain DB and the Desulfosporosinus-dsrA sequence from the SIP incubations have been deposited at GenBank under the accession numbers GU372083 and GU371931, respectively.
Results and discussion
Substrate turnover in peat soil slurry incubations
Six anoxic peat soil slurries were preincubated at 14 °C for 28 days. Electron acceptors such as nitrate, sulfate and iron(III) are typically reduced after 16 days in Schlöppnerbrunnen II peat soil slurries (Küsel et al., 2008), which is an important prerequisite for selective labeling. During this preincubation, lactate, acetate and formate stayed in the lower μM range (<20 μM) whereas propionate accumulated transiently up to 155 μM in individual slurries. Initial sulfate concentrations were 22 μM and dropped thereafter below 4 μM (Figure 1a). After preincubation, an unlabeled substrate mix of lactate, acetate, formate and propionate (50–200 μM each) was added twice over a period of 2 weeks to all soil slurries to determine the time needed for substrate depletion. In addition, three of the six soil slurries were supplemented once with 100–200 μM sulfate. In all incubations, lactate and formate were readily turned over within 2 days, whereas acetate and propionate needed 4 and 6 days for turnover after the first and second substrate addition, respectively (Figure 1b and Table 1). Thereafter, soil slurries were incubated without any additions for 17 days to allow for complete depletion of added 12C-substrates. After this postincubation, the headspace of each mesocosm was flushed with 100% N2 to remove accumulated 12CO2 and the actual SIP incubations were started (preincubations and subsequent SIP incubations are detailed in Supplementary Figure 2).

Substrate and sulfate measurements during preincubation and 12C-substrate turnover determinations. (a) Monitoring of indigenous substrate and sulfate concentrations during 4 weeks of preincubation of anoxic non-amended peat soil slurries. Averages ±s.d. are shown (n=6). (b) Time course of 12C-substrate turnover in anoxic peat soil slurries in the presence and absence of sulfate. Arrows indicate the time points of substrate additions; sulfate was added only once at the beginning of the experiment. Data points represent average values of three independent soil slurries; s.d. bars were omitted for better visibility.
As expected for methanogenic low-sulfate environments, sulfate turnover was slower than substrate consumption, with sulfate reduction accounting for 12% of the total electron flow in incubations with sulfate addition. Sulfate turnover rates as determined by linear regression analysis over the first 9 days were 13.1 μmol SO42− l−1 day−1 (equal to 26.2 nmol SO42− (g soil w. wt.)−1 day−1; Figure 1b) and, thus, in the range of radiotracer-measured in situ sulfate reduction rates of the studied peatland (0 to ca. 340 nmol (g soil w. wt.)−1 day−1; Knorr and Blodau, 2009; Knorr et al., 2009). A possible underestimation of sulfate turnover due to anoxic re-oxidation of sulfide is not expected because of the 28-day-long depletion phase of endogenous electron acceptors before sulfate addition and the observed linearity of sulfate depletion.
A Desulfosporosinus species is the major sulfate reducer in the SIP incubations
To identify active sulfate reducers against the large background of microorganisms involved in methanogenic organic matter degradation, we applied DNA SIP in a differential display format, which involved parallel incubations in the presence or absence of sulfate at in situ concentrations (100–200 μM). Incubations were amended weekly with in situ concentrations of 13C-substrates (composition as for 12C-substrates) with or without sulfate for 2 weeks, 2 months and 6 months—each in a separate mesocosm (six in total). All provided substrates are well known to be used by sulfate reducers. In addition, formate is regarded as an equivalent to H2, which some sulfate reducers use as sole energy source (Rabus et al., 2006). Such autotrophic sulfate reducers can have higher doubling times than heterotrophic sulfate reducers (Rabus et al., 2006) and were targeted in addition by 13CO2, which stemmed from the degradation of the supplied 13C-substrate mixture and was the major source of CO2 available.
Incorporation of substrate-13C into the biomass of active sulfate reducers was followed by pairwise comparison of incubations with and without sulfate using a 16S rRNA gene-based T-RFLP screening of density-resolved DNA. A clear difference in T-RFLP patterns between incubations with and without sulfate became apparent for the bacterial community after 2 months of incubation. In sulfate-amended incubations, a distinct T-RF at 140 bp dominated the ‘heaviest’ (13C-labeled) PCR-amplifiable density fractions and was almost absent in the ‘light’ (unlabeled) fractions. In the control incubation without sulfate, the 140-bp T-RF was of very minor abundance in each fraction throughout the density gradient (Figure 2). The same was true for the 12C-control of non-incubated pristine peat soil (Supplementary Figure 3). Cloning of bacterial 16S rRNA genes from the ‘heavy’ fraction of the incubation with sulfate (Supplementary Table 1 and Supplementary Figure 4) revealed that the dominant 140-bp T-RF represents almost exclusively organisms within the genus Desulfosporosinus (Firmicutes; 16 out of 95 clones in the incubation with sulfate, Figure 3) and one clone of the Acidobacteria subgroup 3. A differential T-RFLP analysis using the alternative restriction enzyme RsaI confirmed that the dominant 140-bp T-RF in the ‘heavy fraction’ represented exclusively Desulfosporosinus spp. (data not shown). Three additional Desulfosporosinus sp. clones had a T-RF at 171 bp (indicating that different Desulfosporosinus ecotypes may be present in the studied peatland) but a corresponding peak was not detected in the SIP–T-RFLP analyses. In a parallel clone library from the heavy fraction of the incubation without sulfate, no Desulfosporosinus sp. was detected (Supplementary Table 1 and Supplementary Figure 4). Again, one clone representing Acidobacteria subgroup 3 with a T-RF of 140 bp was retrieved, explaining the very minor T-RF at 140 bp in the incubations without sulfate (Figure 2) and most likely also in the 12C-control of non-incubated pristine peat soil (Supplementary Figure 3).

T-RFLP fingerprinting of density-resolved bacterial 16S rRNA genes after 2 months of SIP incubations in the presence and absence of sulfate. CsCl buoyant densities are given for each fraction. Major bacterial populations are indicated with their respective T-RFs, which were assigned using 16S rRNA gene clone libraries generated from fractions with buoyant densities 1.722 g ml−1 (incubation with sulfate) and 1.719 g ml−1 (incubation without sulfate), respectively (see also Supplementary Table 1 and Supplementary Figure 4). T-RFs, which had no assignment according to their respective clone library, are indicated by their length only. The range of ‘heavy’ fractions that yielded no PCR product is indicated above the T-RFLP profiles.

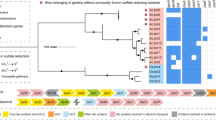
Phylogenetic consensus tree of 16S rRNA gene clones affiliated to the genus Desulfosporosinus (marked in bold). Clones were grouped according to ⩾99% sequence identity; representing T-RFs and number of clones per group are indicated. With one exception, all Desulfosporosinus clones have a 16S rRNA sequence identity of >97% to each other. Parsimony bootstrap values for branches are indicated by solid circles (>90%) and open circles (75–90%). GenBank accession numbers of published 16S rRNA gene sequences are indicated behind the name of the respective sequences. The bar represents 1% estimated sequence divergence as inferred from distance matrix analysis.
The 6-month incubations corroborated that a Desulfosporosinus sp. was the main microorganism that incorporated 13C-label in the presence, but not in the absence, of sulfate (Supplementary Figure 5). Incubating for 2 weeks was apparently too short to detect significant differences between incubations with and without sulfate using in situ substrate concentrations (Supplementary Figure 6). T-RFLP screening of archaeal 16S rRNA genes from the 2-month incubations revealed no differences between density-resolved DNA extracts of incubations with and without sulfate (data not shown), indicating that archaeal sulfate reducers apparently did not have an important role under the conditions tested.
DNA in the ‘heaviest’ PCR-amplifiable density fractions in the 2-month incubations had a density of 1.727 and 1.723 g ml−1 in the incubations with and without sulfate, respectively. This corresponds roughly to 60–70% 13C-labeling based on a G+C content of 50 mol%. In comparison, unlabeled 12C-DNA of microorganisms with a high G+C content such as Micrococcus luteus (G+C content 71 mol%) have a similar buoyant density of up to 1.725 g ml−1 (Lüders et al., 2004). However, all described Desulfosporosinus spp. have a G+C content of 37–47 mol% (Ramamoorthy et al., 2006; Spring and Rosenzweig, 2006; Vatsurina et al., 2008; Lee et al., 2009a; Alazard et al., 2010) and the 140-bp T-RF in the 13C-control without sulfate (Figure 2) as well as in the 12C-control of non-incubated pristine peat soil (Supplementary Figure 3) was of very minor abundance and attributed to Acidobacteria subgroup 3. Therefore, the occurrence of the dominant 140-bp T-RF in the ‘heaviest’ PCR-amplifiable density fraction in the incubation with sulfate clearly reflects 13C-incorporation into Desulfosporosinus sp. In addition, growth on other than the provided, easily degradable 13C-labeled substrates is unlikely because of the 28-day-long preincubation phase before the actual substrate turnover and SIP incubations. A detailed analysis of microbial populations, which were identified in the ‘heaviest’ PCR-amplifiable density fractions of both incubation setups and contributed therefore to metabolic pathways other than sulfate reduction, for example, fermentation in the methanogenic degradation pathway, is given in Supplementary Information.
qPCR analysis confirms physiological activity of the Desulfosporosinus species in the presence of sulfate
DNA replication of Desulfosporosinus sp. in the sulfate-reducing mesocosms was confirmed by quantitative PCR. Although the abundance of Desulfosporosinus sp. in SIP incubations without sulfate mirrored the natural abundance over time (0.006% of total Bacteria and Archaea), it steadily increased to 0.2% (2-week 12C-substrate turnover determination and 2-week SIP incubation), 0.6% (2-week 12C-substrate incubation and 2-month SIP) and 3.1% (2-week 12C-substrate incubation and 6-month SIP) of total bacterial and archaeal 16S rRNA genes in the incubations with sulfate (Figure 4). This result clearly corroborates the observations of the SIP study, which relies on the multiplication of active microorganisms to incorporate label into their DNA, and shows at the same time that the enrichment of Desulfosporosinus sp. in the intensively analyzed 2-month incubation was still minimal.

Quantification of Desulfosporosinus 16S rRNA genes relative to total 16S rRNA genes of Bacteria and Archaea by quantitative real-time PCR. The relative abundance ± s.d. of Desulfosporosinus sp. was determined for pristine peat soil samples over the years 2004, 2006 and 2007 (10–20 cm depth; biological replicates, n=3) in comparison to SIP incubations with and without sulfate (technical replicates, n=3). Peat soil of the 10- to 20-cm depth horizon was also used for the SIP incubations.
The low-abundance Desulfosporosinus can sustain a high cell-specific sulfate reduction rate
Using the onset of our 12C-substrate turnover experiments, in which the number of Desulfosporosinus sp. equaled its natural abundance (also shown by the same abundance in incubations without sulfate, Figure 4), we estimated the cell-specific sulfate reduction rate (cs-SRR) for this Desulfosporosinus sp. to be 341 fmol SO42− cell−1 day−1. The rate was calculated by dividing the measured SRR of the turnover experiments (26.2 nmol SO42− (g soil w. wt.)−1 day−1, Figure 1b) by the abundance of the natural Desulfosporosinus population at 10–20 cm depth. The natural Desulfosporosinus population was estimated from the quantified 16S rRNA gene abundance divided by an average of 4.4 16S rRNA copies per cell among Peptococcaceae for which genome data are available (no data available yet for Desulfosporosinus spp.; Lee et al., 2009b; http://ribosome.mmg.msu.edu/rrndb/index.php). We assumed no significant contribution of additional sulfate reducers to the measured SRR, as Desulfosporosinus sp. was the only recognized sulfate reducer in our SIP incubations. However, activity of other sulfate reducers at the very beginning of our turnover experiments cannot be completely ruled out and thus the determined cs-SRR might be overestimated.
The estimated cs-SRR of the identified peatland Desulfosporosinus sp. is at the upper end of cs-SRRs reported for pure cultures (Detmers et al., 2001). In marine sediments, cs-SRRs are three orders of magnitude lower, which is explained by substrate limitation (Sahm et al., 1999; Ravenschlag et al., 2000). The studied peatland, however, is not regarded as substrate limited, which is supported by long periods of a high dissolved organic carbon content (average 18 mg l−1) and the spatial and temporal co-occurrence of redox processes with differing energy yield (Alewell et al., 2008; Küsel et al., 2008). In addition, apparent sulfate half-saturation concentrations, Km, for sulfate reducers from low-sulfate environments can be as low as 5 μM, indicating no kinetic limitation from the electron acceptor side as well (Pallud and van Cappellen, 2006, and references therein). This is supported by the high radiotracer-measured SRRs of the studied peatland even at bulk sulfate concentrations of <10 μM (Knorr and Blodau, 2009; Knorr et al., 2009).
An intriguing question remaining is whether sulfate supply might hamper the small Desulfosporosinus population to sustain such high cs-SRR. In an extreme scenario, each cell of the highly diluted Desulfosporosinus population would rapidly turn over sulfate in its close vicinity and, thereafter, might run into sulfate limitation controlled by diffusion of sulfate to the cell. To test this, we calculated the diffusive flux J of sulfate to a single Desulfosporosinus cell using a diffusion coefficient for sulfate as determined experimentally for anoxic sediments (DS=0.5 × 10−5 cm2 s−1; Krom and Berner, 1980), a cell radius r for Desulfosporosinus cells of 0.4 μM (Spring and Rosenzweig, 2006), and a three-dimensional diffusive flux model for spherical symmetries (Equation 1; Koch, 1990).

Using a low ambient sulfate concentration cmax in the peat of 10 μM (typically 10–300 μM), the diffusive flux into a single Desulfosporosinus cell would be 2 pmol SO42− day−1. Even with this conservative estimate, the diffusive flux of sulfate would be one order of magnitude higher than the estimated cs-SRR of Desulfosporosinus sp. in situ (341 fmol SO42− cell−1 day−1). This clearly shows that Desulfosporosinus cells or any other sulfate reducer will not be limited by sulfate diffusion and make such high cs-SRR also plausible in the natural peatland.
The low-abundance Desulfosporosinus has the potential to drive a substantial part of sulfate reduction in the peatland
Desulfosporosinus sp., with its low natural abundance of 0.006% of the total bacterial and archaeal community, is a member of the ‘rare biosphere’, which is defined as the sum of those taxa with an abundance of <0.1–1% (Pedros-Alio, 2006; Sogin et al., 2006; Fuhrman, 2009). Based on its absolute abundance in the peatland over a depth profile of 0–30 cm (Figure 5), the potential SRR of the natural Desulfosporosinus population was calculated using its estimated cs-SRR. The potential SRRs of the natural Desulfosporosinus population were 4.0–36.8 nmol (g soil w. wt.)−1 day−1 between 0 and 30 cm soil depth (Figure 5). In comparison, radiotracer-measured gross SRRs of the studied peatland ranged from 0 to ca. 340 nmol (g soil w. wt.)−1 day−1 over a depth profile of 0–30 cm and a 300-day period, with sulfate reduction proceeding at >10 nmol (g soil w. wt.)−1 day−1 in at least one of the analyzed depth fractions at each sampling day (5–10 cm depth fractions; Knorr and Blodau, 2009; Knorr et al., 2009). Even if cs-SRRs of Desulfosporosinus sp. were overestimated by one order of magnitude and would therefore resemble average cs-SRRs of cultured sulfate reducers (Detmers et al., 2001) or if a subpopulation would have occurred as inactive spores, the natural Desulfosporosinus population would still have the potential to drive a considerable part of sulfate reduction compared with its abundance. The presence of mostly physiologically active Desulfosporosinus cells in water-saturated, anoxic soil pockets above the water table and in the anoxic peat below the water table is expected, as sulfate reduction in peatlands is not only fuelled by allochthonous sulfate but also by an oxic (Deppe et al., 2009; Knorr and Blodau, 2009; Knorr et al., 2009; Reiche et al., 2009) and anoxic sulfur cycle (Jørgensen, 1990b; Blodau et al., 2007; Nielsen et al., 2010) and constitutes an ongoing process in the studied peatland, as evident from δ34S measurements (see, for example, Alewell and Novak, 2001; Alewell et al., 2008) and the radiotracer studies described above. In addition, Desulfosporosinus spp. are known to switch under sulfate limitation to the fermentation of lactate and pyruvate (Spring and Rosenzweig, 2006), to reductive acetogenesis from formate, methanol or methyl groups of aromatic compounds (Rabus et al., 2006) or to dissimlatory iron(III) reduction (Ramamoorthy et al., 2006). At the same time, Desulfosporosinus spp. are well adapted to persist throughout extended periods of droughts and subsequent complete oxygenation of the peat soil (Reiche et al., 2009) by their ability to form endospores (Ramamoorthy et al., 2006; Spring and Rosenzweig, 2006; Vatsurina et al., 2008; Lee et al., 2009a). In summary, Desulfosporosinus spp. seem well adapted to the highly fluctuating conditions in low-sulfate peatlands.

Quantification of Desulfosporosinus 16S rRNA gene numbers by quantitative PCR over a peatland depth profile of 0–30 cm and quantification of potential Desulfosporosinus sulfate reduction rates (SRRs). 16S rRNA gene numbers were determined in triplicate cores over the years 2004, 2006 and 2007, with the exception of the 20- to 30-cm depth, in which samples were only available for the year 2007. The distribution of gene numbers is represented in boxplots showing the interquartile range and the median. Whiskers (maximum 1.5-fold interquartile range) represent the data distribution outside the interquartile range; outliers are depicted as black circles. Potential SRRs of the Desulfosporosinus population were determined using the estimated cell-specific SRR of the identified peatland Desulfosporosinus sp., the interquartile range of Desulfosporosinus 16S rRNA genes per depth and an average of 4.4 16S rRNA gene copies per cell (for details, see text).
dsrAB-based SIP identifies no substantial contribution of other sulfate reducers
As evident from gross SRRs (Knorr and Blodau, 2009; Knorr et al., 2009) and previous diversity studies, several other sulfate reducers are present in this peatland, for example, Desulfomonile spp. and Syntrophobacter spp. (Loy et al., 2004). In addition, the presence of potentially new taxa is indicated by the detection of novel deep-branching lineages of the functional marker genes dsrAB (Loy et al., 2004; Schmalenberger et al., 2007). Currently, it is not known whether microorganisms harboring these novel dsrAB are capable of dissimilatory sulfate/sulfite (Wagner et al., 2005) or organosulfonate reduction (Laue et al., 1997, 2001), switch between a syntrophic and sulfate-reducing lifestyle upon the availability of sulfate (Wallrabenstein et al., 1994, 1995) or are purely syntrophic microorganisms (Imachi et al., 2006). Comparison of dsrAB clone libraries from the ‘heavy’ fractions of the SIP incubations with and without sulfate (Figure 6 and Table 2) and dsrAB T-RFLP analyses for both incubations (Supplementary Figure 7) indicated that known sulfate reducers other than Desulfosporosinus or microorganisms harboring novel deep-branching dsrAB made no quantitatively important contribution (if any) to sulfate reduction (detailed in Supplementary Information). However, it is very likely that additional sulfate reducers in the studied peatland use other substrates for energy metabolism or are adapted to other conditions than those provided in our SIP incubations and, thus, were not identified as active populations.

Phylogenetic consensus tree of deduced DsrAB amino acid sequences longer than 500 amino acids, showing the affiliation of operational taxonomical units (OTUs) retrieved from the ‘heavy’ SIP fractions (indicated by a triangle) in comparison to known sulfate reducers and peat soil OTUs retrieved in a previous study from the same and a neighboring peatland (Loy et al., 2004). An OTU comprises all sequences having ⩾90% amino acid sequence identity. Deduced DsrAB sequences shorter than 500 amino acids (indicated by dashed branches) were individually added to the distance matrix tree without changing the overall tree topology using the ARB Parsimony_interactive tool (Ludwig et al., 2004). Parsimony bootstrap values for branches are indicated by solid circles (>90%) and open circles (75–90%). The Desulfosporosinus-related dsrA clone (shown in red) was retrieved from the 2-month incubation with sulfate using Desulfosporosinus/Desulfitobacterium-selective primers. GenBank accession numbers of published DsrAB sequences are indicated behind the name of the respective sequences. The bar represents 10% estimated sequence divergence as inferred from distance matrix analysis.
Interestingly, Desulfosporosinus-like dsrAB could not be detected in the ‘heavy’ fractions when the standard highly degenerated primers were applied for PCR, indicating that the labeled Desulfosporosinus sp. harbors dsrAB with mismatches in the primer binding sites, as observed previously also for other closely related sulfate reducers (Zverlov et al., 2005). Consistent with this hypothesis, a dsrA fragment closely related to dsrAB of Desulfosporosinus spp. (Figure 6) and probably related to one of the detected Desulfosporosinus ecotypes (Figure 3) could be amplified after using newly constructed primers targeting selectively the genera Desulfosporosinus and Desulfitobacterium. This also explains why this ‘rare biosphere’ member eluded previous dsrAB-based diversity studies of this peatland.
Conclusions
Microbial diversity surveys using high-throughput sequencing of 16S rRNA gene amplicons revealed that any microbial community in the environment is typically composed of abundant taxa, which are considered to carry out most ecosystem functions, and very low abundant taxa (<0.1–1%), which are referred to as the ‘rare biosphere’ (Pedros-Alio, 2006; Sogin et al., 2006; Roesch et al., 2007; Fuhrman, 2009; Turnbaugh et al., 2009; Webster et al., 2010). Recent advances in data analyses allow more precise estimates of the actual extent of the ‘rare biosphere’ diversity (Kunin et al., 2009; Quince et al., 2009). However, we are only starting to learn what different ecological roles these rare microorganisms may have. In this study, we present cumulative and independent lines of evidence that a Desulfosporosinus species as member of the ‘rare biosphere’ is an important sulfate reducer in the investigated model peatland. This finding provides an example for a microorganism of numerical low abundance that can have an impact on (1) the carbon flow in terrestrial ecosystems (Vile et al., 2003; Loy et al., 2004; Blodau et al., 2007; this study) and (2) on globally relevant processes such as the decrease in emission of the greenhouse gas methane (Gauci et al., 2004). A similar situation has been recently observed for marine and freshwater environments. In coastal marine surface waters, a low-abundant Methylophaga sp. was shown to be the major methanol oxidizer (Neufeld et al., 2008), and in the chemocline of a meromictic lake, a low abundance but large anaerobic phototrophic bacterium (0.1–0.4% of total cells) accounted for 40% of ammonium and 70% of inorganic carbon uptake (Musat et al., 2008; Halm et al., 2009). However, in contrast to our study the latter was explained by the large biomass (40% of the total microbial biomass) of these voluminous lake bacteria (Musat et al., 2008; Halm et al., 2009).
Two major advantages of a low-abundance life strategy have been outlined before: protection against viral lysis and protection against protist predation due to lowered probabilities of encounters (Pedros-Alio, 2006). In addition, the low abundance of the peatland Desulfosporosinus sp. may be caused by the observed fluctuating conditions in peatlands such as de-coupled variations in substrate and sulfate concentrations (Küsel et al., 2008), irregularly occurring disturbance events such as oxygen exposure by droughts or heavy rainfall (Deppe et al., 2009; Knorr and Blodau, 2009; Knorr et al., 2009; Reiche et al., 2009) and continuous energy consumption for maintenance, for example, by simultaneous sulfate reduction and oxygen detoxification at oxic–anoxic interfaces (Brune et al., 2000) or by maintaining intracellular pH homeostasis in the acidic peatland. Such stresses were partially relieved in our SIP incubations, leading to a possible re-channeling of energy requirements from maintenance toward growth, and are a possible explanation for the observed slow increase of the Desulfosporosinus population over the incubation period. Alternatively, the identified peatland Desulfosporosinus might represent an r-strategist making use of the slightly elevated sulfate concentrations (100–300 μM) during and after oxygenation events, which often occur in the analyzed 10–20 cm depth fraction due to water table fluctuations. At the same time, it would have to cope with oxygen and pH stress, again restricting its energy supply for growth. Interestingly, gross in situ SRRs during such oxygenation events increase several-fold reaching peak rates of >600 nmol (g soil w. wt.)−1 day−1, which is explained by sulfate reduction in water-saturated, anoxic soil pockets above the water table (Knorr and Blodau, 2009; Knorr et al., 2009).
Our findings highlight that microbial communities do not only consist of abundant microorganisms, which carry out the major ecosystem functions, and the dormant ‘rare biosphere’, which results from random dispersal and/or functions as a ‘microbial seed bank’ and insurance for the case of changing environmental conditions (Pedros-Alio, 2006; Fuhrman, 2009; Hubert et al., 2009; Patterson, 2009; Turnbaugh et al., 2009). In addition, microbial keystone species, such as Desulfosporosinus sp. in the peatland, ‘whose effect is large and disproportionately large relative to their abundance’ (Power et al., 1996), are apparently of considerable importance in certain ecosystems.
Accession codes
References
Alazard D, Joseph M, Battaglia-Brunet F, Cayol J-L, Ollivier B . (2010). Desulfosporosinus acidiphilus sp. nov.: a moderately acidophilic sulfate-reducing bacterium isolated from acid mining drainage sediments. Extremophiles 14: 305–312.
Alewell C, Novak M . (2001). Spotting zones of dissimilatory sulfate reduction in a forested catchment: the 34S-35S approach. Environ Poll 112: 369–377.
Alewell C, Paul S, Lischeid G, Storck FR . (2008). Co-regulation of redox processes in freshwater wetlands as a function of organic matter availability? Sci Total Environ 404: 335–342.
Berge E, Bartnicki J, Olendrzynski K, Tsyro SG . (1999). Long-term trends in emissions and transboundary transport of acidifying air pollution in Europe. J Environ Manag 57: 31–50.
Blodau C, Mayer B, Peiffer S, Moore TR . (2007). Support for an anaerobic sulfur cycle in two Canadian peatland soils. J Geophys Res-Biogeosci 112: 1–10.
Bodrossy L, Stralis-Pavese N, Konrad-Koszler M, Weilharter A, Reichenauer TG, Schofer D et al. (2006). mRNA-based parallel detection of active methanotroph populations by use of a diagnostic microarray. Appl Environ Microbiol 72: 1672–1676.
Brune A, Frenzel P, Cypionka H . (2000). Life at the oxic-anoxic interface: microbial activities and adaptations. FEMS Microbiol Rev 24: 691–710.
Deppe M, McKnight DM, Blodau C . (2009). Effects of short-term drying and irrigation on electron flow in mesocosms of a northern bog and an alpine fen. Environ Sci Technol 44: 80–86.
Detmers J, Bruchert V, Habicht KS, Kuever J . (2001). Diversity of sulfur isotope fractionations by sulfate-reducing prokaryotes. Appl Environ Microbiol 67: 888–894.
Fuhrman JA . (2009). Microbial community structure and its functional implications. Nature 459: 193–199.
Gauci V, Matthews E, Dise N, Walter B, Koch D, Granberg G et al. (2004). Sulfur pollution suppression of the wetland methane source in the 20th and 21st centuries. Proc Natl Acad Sci USA 101: 12583–12587.
Halm H, Musat N, Lam P, Langlois R, Musat F, Peduzzi S et al. (2009). Co-occurrence of denitrification and nitrogen fixation in a meromictic lake, Lake Cadagno (Switzerland). Environ Microbiol 11: 1945–1958.
Heitmann T, Blodau C . (2006). Oxidation and incorporation of hydrogen sulfide by dissolved organic matter. Chem Geol 235: 12–20.
Heitmann T, Goldhammer T, Beer J, Blodau C . (2007). Electron transfer of dissolved organic matter and its potential significance for anaerobic respiration in a northern bog. Glob Change Biol 13: 1771–1785.
Houweling S, Kaminski T, Dentener F, Lelieveld J, Heimann M . (1999). Inverse modeling of methane sources and sinks using the adjoint of a global transport model. J Geophys Res-Atmos 104: 26137–26160.
Hubert C, Loy A, Nickel M, Arnosti C, Baranyi C, Bruchert V et al. (2009). A constant flux of diverse thermophilic bacteria into the cold Arctic seabed. Science 325: 1541–1544.
Imachi H, Sekiguchi Y, Kamagata Y, Loy A, Qiu YL, Hugenholtz P et al. (2006). Non-sulfate-reducing, syntrophic bacteria affiliated with Desulfotomaculum cluster I are widely distributed in methanogenic environments. Appl Environ Microbiol 72: 2080–2091.
Jørgensen BB . (1990a). A thiosulfate shunt in the sulfur cycle of marine sediments. Science 249: 152–154.
Jørgensen BB . (1990b). The sulfur cycle of fresh-water sediments—role of thiosulfate. Limnol Oceanogr 35: 1329–1342.
Knorr K-H, Blodau C . (2009). Impact of experimental drought and rewetting on redox transformations and methanogenesis in mesocosms of a northern fen soil. Soil Biol Biochem 41: 1187–1198.
Knorr KH, Lischeid G, Blodau C . (2009). Dynamics of redox processes in a minerotrophic fen exposed to a water table manipulation. Geoderma 153: 379–392.
Koch AL . (1990). Diffusion—the crucial process in many aspects of the biology of bacteria. Adv Microb Ecol 11: 37–70.
Krom MD, Berner RA . (1980). The diffusion-coefficients of sulfate, ammonium, and phosphate ions in anoxic marine sediments. Limnol Oceanogr 25: 327–337.
Kunin V, Engelbrektson A, Ochman H, Hugenholtz P . (2009). Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol 12: 118–123.
Küsel K, Blothe M, Schulz D, Reiche M, Drake HL . (2008). Microbial reduction of iron and porewater biogeochemistry in acidic peatlands. Biogeosciences 5: 1537–1549.
Laue H, Denger K, Cook AM . (1997). Taurine reduction in anaerobic respiration of Bilophila wadsworthia RZATAU. Appl Environ Microbiol 63: 2016–2021.
Laue H, Friedrich M, Ruff J, Cook AM . (2001). Dissimilatory sulfite reductase (desulfoviridin) of the taurine-degrading, non-sulfate-reducing bacterium Bilophila wadsworthia RZATAU contains a fused DsrB-DsrD subunit. J Bacteriol 183: 1727–1733.
Lee Y-J, Romanek CS, Wiegel J . (2009a). Desulfosporosinus youngiae sp. nov., a spore-forming, sulfate-reducing bacterium isolated from a constructed wetland treating acid mine drainage. Int J Syst Evol Microbiol 59: 2743–2746.
Lee ZM-P, Bussema III C, Schmidt TM . (2009b). rrnDB: documenting the number of rRNA and tRNA genes in bacteria and archaea. Nucl Acids Res 37: D489–D493.
Limpens J, Berendse F, Blodau C, Canadell JG, Freeman C, Holden J et al. (2008). Peatlands and the carbon cycle: from local processes to global implications a synthesis. Biogeosciences 5: 1475–1491.
Loy A, Küsel K, Lehner A, Drake HL, Wagner M . (2004). Microarray and functional gene analyses of sulfate-reducing prokaryotes in low-sulfate, acidic fens reveal cooccurrence of recognized genera and novel lineages. Appl Environ Microbiol 70: 6998–7009.
Loy A, Lehner A, Lee N, Adamczyk J, Meier H, Ernst J et al. (2002). Oligonucleotide microarray for 16S rRNA gene-based detection of all recognized lineages of sulfate-reducing prokaryotes in the environment. Appl Environ Microbiol 68: 5064–5081.
Lüders T, Manefield M, Friedrich MW . (2004). Enhanced sensitivity of DNA- and rRNA-based stable isotope probing by fractionation and quantitative analysis of isopycnic centrifugation gradients. Environ Microbiol 6: 73–78.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar et al. (2004). ARB: a software environment for sequence data. Nucl Acids Res 32: 1363–1371.
Matzner E . (2004). Biogeochemistry of Forested Catchments in a Changing Environment—a German Case Study, vol. 172. Springer Verlag: Berlin.
Moldan B, Schnoor JL . (1992). Czechoslovakia—examining a critically ill environment. Environ Sci Technol 26: 14–21.
Musat N, Halm H, Winterholler B, Hoppe P, Peduzzi S, Hillion F et al. (2008). A single-cell view on the ecophysiology of anaerobic phototrophic bacteria. Proc Natl Acad Sci USA 105: 17861–17866.
Neufeld JD, Chen Y, Dumont MG, Murrell JC . (2008). Marine methylotrophs revealed by stable-isotope probing, multiple displacement amplification and metagenomics. Environ Microbiol 10: 1526–1535.
Neufeld JD, Vohra J, Dumont MG, Lüders T, Manefield M, Friedrich MW et al. (2007). DNA stable-isotope probing. Nat Protoc 2: 860–866.
Nielsen LP, Risgaard-Petersen N, Fossing H, Christensen PB, Sayama M . (2010). Electric currents couple spatially separated biogeochemical processes in marine sediment. Nature 463: 1071–1074.
Pallud C, van Cappellen P . (2006). Kinetics of microbial sulfate reduction in estuarine sediments. Geochim Cosmochim Acta 70: 1148–1162.
Patterson DJ . (2009). Seeing the big picture on microbe distribution. Science 325: 1506–1507.
Paul S, Küsel K, Alewell C . (2006). Reduction processes in forest wetlands: tracking down heterogeneity of source/sink functions with a combination of methods. Soil Biol Biochem 38: 1028–1039.
Pedros-Alio C . (2006). Marine microbial diversity: can it be determined? Trends Microbiol 14: 257–263.
Power ME, Tilman D, Estes JA, Menge BA, Bond WJ, Mills LS et al. (1996). Challenges in the quest for keystones. BioScience 46: 609–620.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucl Acids Res 35: 7188–7196.
Quince C, Lanzen A, Curtis TP, Davenport RJ, Hall N, Head IM et al. (2009). Accurate determination of microbial diversity from 454 pyrosequencing data. Nat Methods 6: 639–641.
Rabus R, Hansen TA, Widdel F . (2006). Dissimilatory sulfate- and sulfur-reducing prokaryotes. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (eds). The Prokaryotes: A Handbook on the Biology of Bacteria, 3 edn. Springer: Singapore. pp 659–768.
Ramamoorthy S, Sass H, Langner H, Schumann P, Kroppenstedt RM, Spring S et al. (2006). Desulfosporosinus lacus sp nov., a sulfate-reducing bacterium isolated from pristine freshwater lake sediments. Int J Syst Evol Microbiol 56: 2729–2736.
Rasch PJ, Tilmes S, Turco RP, Robock A, Oman L, Chen CC et al. (2008). An overview of geoengineering of climate using stratospheric sulphate aerosols. Philos Trans R Soc A-Math Phys Eng Sci 366: 4007–4037.
Ravenschlag K, Sahm K, Knoblauch C, Jørgensen BB, Amann R . (2000). Community structure, cellular rRNA content, and activity of sulfate-reducing bacteria in marine Arctic sediments. Appl Environ Microbiol 66: 3592–3602.
Reiche M, Hadrich A, Lischeid G, Küsel K . (2009). Impact of manipulated drought and heavy rainfall events on peat mineralization processes and source-sink functions of an acidic fen. J Geophys Res-Biogeosci 114: G02021, doi:02010.01029/02008jg000853.
Roesch LFW, Fulthorpe RR, Riva A, Casella G, Hadwin AKM, Kent AD et al. (2007). Pyrosequencing enumerates and contrasts soil microbial diversity. ISMEJ 1: 283–290.
Sahm K, MacGregor BJ, Jørgensen BB, Stahl DA . (1999). Sulphate reduction and vertical distribution of sulphate-reducing bacteria quantified by rRNA slot-blot hybridization in a coastal marine sediment. Environ Microbiol 1: 65–74.
Schmalenberger A, Drake HL, Küsel K . (2007). High unique diversity of sulfate-reducing prokaryotes characterized in a depth gradient in an acidic fen. Environ Microbiol 9: 1317–1328.
Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR et al. (2006). Microbial diversity in the deep sea and the underexplored ‘rare biosphere’. Proc Natl Acad Sci USA 103: 12115–12120.
Spring S, Rosenzweig F . (2006). The genera Desulfitobacterium and Desulfosporosinus: taxonomy. In: Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E (eds). The Prokaryotes: A Handbook on the Biology of Bacteria, 3 edn. Springer: Singapore. pp 771–786.
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE et al. (2009). A core gut microbiome in obese and lean twins. Nature 457: 480–484.
Vatsurina A, Badrutdinova D, Schumann P, Spring S, Vainshtein M . (2008). Desulfosporosinus hippei sp. nov., a mesophilic sulfate-reducing bacterium isolated from permafrost. Int J Syst Evol Microbiol 58: 1228–1232.
Vile MA, Bridgham SD, Wieder RK, Novak M . (2003). Atmospheric sulfur deposition alters pathways of gaseous carbon production in peatlands. Global Biogeochem Cycles 17, doi: 10.1029/2002gb001966.
Wagner M, Loy A, Klein M, Lee N, Ramsing NB, Stahl DA et al. (2005). Functional marker genes for identification of sulfate-reducing prokaryotes. Environmental Microbiology. Elsevier Academic Press Inc: San Diego. pp 469–489.
Wallrabenstein C, Hauschild E, Schink B . (1994). Pure culture and cytological properties of Syntrophobacter wolinii. FEMS Microbiol Lett 123: 249–254.
Wallrabenstein C, Hauschild E, Schink B . (1995). Syntrophobacter pfennigii sp nov, new syntrophically propionate-oxidizing anaerobe growing in pure culture with propionate and sulfate. Arch Microbiol 164: 346–352.
Webster NS, Taylor MW, Behnam F, Lücker S, Rattei T, Whalan S et al. (2010). Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ Microbiol: doi: 10.1111/j.1462-2920.2009.02065.x.
Wind T, Conrad R . (1997). Localization of sulfate reduction in planted and unplanted rice field soil. Biogeochemistry 37: 253–278.
Wuebbles DJ, Hayhoe K . (2002). Atmospheric methane and global change. Earth-Sci Rev 57: 177–210.
Zverlov V, Klein M, Lucker S, Friedrich MW, Kellermann J, Stahl DA et al. (2005). Lateral gene transfer of dissimilatory (bi)sulfite reductase revisited. J Bacteriol 187: 2203–2208.
Acknowledgements
This research was financially supported by the Alexander-von-Humboldt-Foundation (MP), the Austrian Science Fund (P18836-B17 and P20185-B17, AL), the German Federal Ministry of Science and Education (BIOLOG/BIOTA project 01LC0621D; MW and AL), the Austrian Federal Ministry of Science and Research (ASEA-UNINET; PD) and the University of Vienna (PD). We gratefully acknowledge the technical assistance of Christian Baranyi and Stefan Flechl and thank Casey Hubert and David Berry for critical comments on the paper, Olga V. Karnachuk for Desulfosporosinus strain DB cell material and Andreas Richter for 13C-substrate quality analysis and the possibility to use the IC-HPLC.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Pester, M., Bittner, N., Deevong, P. et al. A ‘rare biosphere’ microorganism contributes to sulfate reduction in a peatland. ISME J 4, 1591–1602 (2010). https://doi.org/10.1038/ismej.2010.75
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2010.75
Keywords
This article is cited by
-
Lignocellulosic fraction-induced niche differentiation within dissimilatory iron reducing bacterial groups in a paddy soil
Soil Ecology Letters (2024)
-
The different responses of planktonic bacteria and archaea to water temperature maintain the stability of their community diversity in dammed rivers
Ecological Processes (2023)
-
Antimicrobial peptides act on the rumen microbiome and metabolome affecting the performance of castrated bulls
Journal of Animal Science and Biotechnology (2023)
-
The selection of copiotrophs may complicate biodiversity-ecosystem functioning relationships in microbial dilution-to-extinction experiments
Environmental Microbiome (2023)
-
Oxygen respiration and polysaccharide degradation by a sulfate-reducing acidobacterium
Nature Communications (2023)
