
Abstract
Microbial compositions of human and animal feces from South Korea were analyzed and characterized. In total, 38 fecal samples (14 healthy adult humans, 6 chickens, 6 cows, 6 pigs and 6 geese) were analyzed by 454 pyrosequencing of the V2 region of the 16S rRNA gene. Four major phyla, Actinobacteria, Proteobacteria, Firmicutes and Bacteroidetes, were identified in the samples. Principal coordinate analysis suggested that microbiota from the same host species generally clustered, with the exception of those from humans, which exhibited sample-specific compositions. A network-based analysis revealed that several operational taxonomic units (OTUs), such as Lactobacillus sp., Clostridium sp. and Prevotella sp., were commonly identified in all fecal sources. Other OTUs were present only in fecal samples from a single organism. For example, Yania sp. and Bifidobacterium sp. were identified specifically in chicken and human fecal samples, respectively. These specific OTUs or their respective biological markers could be useful for identifying the sources of fecal contamination in water by microbial source tracking.
Similar content being viewed by others

Main
Microbial source tracking (MST) is currently receiving increasing attention for its use in prevention of water contamination and in accurate assessment of human health risks. MST is a tool used to identify the origin of fecal contamination in water through various molecular and biochemical methods (Simpson et al., 2002; Seurinck et al., 2005; Gourmelon et al., 2007). Target microorganisms for MST typically include conventional fecal indicator microorganisms, including Escherichia coli, enterococci and bacteriophages (Scott et al., 2002). Traditionally, these target microorganisms are isolated from samples, and various genotypic and/or phenotypic methods are applied to properly categorize the fecal origin of the microorganisms. Recently, culture-independent molecular markers, such as the 16S rRNA gene or taxon-specific genes (for example, the Enterococcus spp. esp gene and the Bacteroidales-specific 16S rRNA gene), have been identified and used for MST analysis (Fogarty and Voytek, 2005; King et al., 2007; Byappanahalli et al., 2008). However, use of this technique is limited by the fact that not all of these molecular or biochemical markers can provide the correct identification of fecal sources.
Comparison of fecal microbiota represents another method that could be used for the identification of the most likely source organism in MST analysis. Recently, 454 pyrosequencing has become a powerful tool that can be used to analyze millions of nucleic acid sequences with a low level of error (<1%) to determine microbiota composition (Margulies et al., 2005; Andersson et al., 2008). However, to date, few studies have characterized the fecal microbiota in human and animal feces (Andersson et al., 2008; Turnbaugh et al., 2009), and this method has not been used for MST. Thus, the objectives of this study were to characterize the compositions of the microbial communities in human and animal feces to identify specific microorganisms in each host for potential use in MST analysis.
In total, 38 fecal samples, including 14 human, 6 chicken, 6 cow, 6 pig and 6 wild geese, were collected for this study. Human fecal samples were obtained from healthy adults with normal body mass indices. Livestock fecal samples were collected from various farms located in Kyunggi Province, South Korea as described in a previous study (Lee et al., 2009). Fecal samples from wild geese were collected from their habitats in the Cheonra area of the southeastern part of South Korea.
Total DNA was extracted from feces using previously described methods (Turnbaugh et al., 2009). The 16S rRNA gene was then amplified from isolated DNA using a previously reported primer set (Fierer et al., 2008). The 27F and 338R primers were used as forward and reverse primers and contained the 454 Life Sciences (Branford, CT, USA) primers B and A, respectively. A unique 5-bp error-correcting bar code was used to tag each PCR product. Amplified PCR products were purified using the QIAquick PCR Purification Kit (Qiagen, Valencia, CA, USA). The resulting samples were sent to the National Instrumentation Center for Environmental Management at Seoul National University for pyrosequencing using a 454 Life Sciences Genome Sequencer FLX machine (Roche, Basel, Switzerland).
Sequences were processed and analyzed as previously described (Fierer et al., 2008; Ley et al., 2008). For each of the 38 fecal samples, 3000 reads of the V2 region were randomly selected. The nucleic acid sequences of the 16S rRNA gene were trimmed using the Greengenes database with a quality score of <20. A multiple sequence alignment was generated using NAST (parameters: minimum alignment length, 200 bp; sequence identity, 70%) (DeSantis et al., 2006a). The taxonomy of each phylotype was classified based on the Greengenes database (DeSantis et al., 2006b) using the Ribosomal Database Project taxonomy. Pre-aligned sequences were added to a neighbor-joining tree available from the Greengenes core set database using ARB project (http://www.arb-home.de). Then, the UniFrac program was used to conduct principal coordinate analysis (Lozupone and Knight, 2005). Finally, a network-based analysis using Cytoscape 2.6.3 was performed to identify core and specific operational taxonomic units in each host (Shannon et al., 2003).
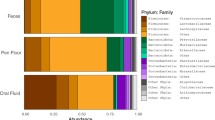
The V2 region of the 16S rRNA gene was pyrosequenced with an average of 7252±2107 reads per sample, and 3000 reads were randomly selected for each sample and subjected to analysis as described in a previous study (Turnbaugh et al., 2009). The vast majority (>99%) of classified sequences belonged to four phyla, Firmicutes, Bacteroidetes, Actinobacteria and Proteobacteria (Supplementary Figure S1). When the phylogenetic characteristics of the gut microbiota in each host were compared based on the average unweighted UniFrac distance, the human gut microbiota was found to be significantly different from that of all other animals. The microbiota in chicken fecal matter exhibited the greatest difference from human fecal samples (***P<10−6), and the microbiota in pig samples exhibited the least difference from human fecal samples (*P<0.01) (Supplementary Figure S3). The Shannon–Weaver indices were highest in cows (4.53), followed by humans (4.14), chickens (4.02), pigs (3.90) and geese (3.67) (Supplementary Table S1, Supplementary Figure S2).
Principal coordinate analysis revealed clustering based on the origin of the fecal samples (Figure 1a), with the exception of human samples. Human samples were found to be scattered throughout the plot. When principal coordinate analysis was analyzed for each of the phyla represented, members of the Firmicutes phylum were found to be primarily responsible for the clustering observed in the principal coordinate analysis (Supplementary Figure S4), indicating that bacteria belonging to this phylum may represent useful microbiological and molecular targets for MST.

Clustering of fecal bacterial communities by principal coordinate analysis (PCoA) using UniFrac (• chicken, ▴ cow, ▪ goose, ♦ pig and – human) (a) and results from network-based analysis of microbiota (b and c) (N=38). Nodes represent operational taxonomic units (OTUs), and each line indicates that an OTU was identified in the same source.
Our results indicate that the general compositions of the gut microbiota in humans and other vertebrates were similar overall (Supplementary Table S2). However, specific differences, such as alterations in microbial diversity and the presence of specific microorganisms, were identified in each of the gut microbiota samples analyzed. Several previous studies have suggested that high levels of variation are present in the gut microbiota of different populations (Andersson et al., 2008; Ley et al., 2008). In particular, the gut microbiota composition can be affected by various factors, including diet, age and obesity (Ley et al., 2006). For these reasons, body mass index and age in human subjects were carefully controlled in this study.
Figures 1b and c depict a network-based analysis of fecal microbiota obtained using the Cytoscape program (Ley et al., 2008). This analysis revealed that microbial communities from the same fecal origin were highly similar (Figure 1b). In addition to a common core microbiota, several microorganisms were found specifically in samples of a particular fecal origin (Figure 1c). For example, Bifidobacterium spp. were identified only in human samples (21% of analyzed samples). Therefore, Bifidobacterium could be considered to be a specific fecal microorganism representing fecal contamination from humans. These results were consistent with previous studies (Long et al., 2005; Dorai-Raj et al., 2009). In addition, Yania spp. were found in 33% of chicken fecal samples and were specifically identified only in chickens (Table 1). These results suggest that Yania could be useful as a specific indicator bacterium for fecal contamination from chickens. Other operational taxonomic units specific to fecal origin included Agromyces spp. (goose) and Marinicola spp. (pig). The presence of these host-specific microorganisms could be because of various biological and ecological characteristics, such as diet and environmental exposure (Turnbaugh et al., 2009), and may be useful as host-specific biomarkers for MST. However, future studies assessing the use of these markers for MST should characterize the fate and transport of these fecal microorganisms in a water-based environment, as this study analyzed only fecal samples. In conclusion, we have characterized and identified specific microbiota in human and animal fecal samples. Further studies using a larger sample size representative of different geographic regions should be performed in the future to better characterize the distributions of differences in microbiota across a variety of species.
References
Andersson AF, Lindberg M, Jakobsson H, Bäckhed F, Nyrén P, Engstrand L . (2008). Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 3: e2836.
Byappanahalli MN, Przybyla-Kelly K, Shively DA, Whitman RL . (2008). Environmental occurrence of the enterococcal surface protein (esp) gene is an unreliable indicator of human fecal contamination. Environ Sci Technol 42: 8014–8020.
DeSantis TZ, Hugenholtz P, Keller K, Brodie EL, Larsen N, Piceno YM et al. (2006a). NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res 34: W394–W399.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K et al. (2006b). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72: 5069–5072.
Dorai-Raj S, Grady JO, Colleran E . (2009). Specificity and sensitivity evaluation of novel and existing Bacteroidales and Bifidobacteria-specific PCR assays on feces and sewage samples and their application for microbial source tracking in Ireland. Water Res 43: 4980–4988.
Fierer N, Hamady M, Lauber CL, Knight R . (2008). The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci USA 105: 17994–17999.
Fogarty L, Voytek M . (2005). Comparison of Bacteroides-Prevotella 16S rRNA genetic markers for fecal samples from different animal species. Appl Environ Microbiol 71: 5999–6007.
Gourmelon M, Caprais MP, Segura R, Le Mennec C, Lozach S, Piriou JY et al. (2007). Evaluation of two library-independent microbial source tracking methods to identify sources of fecal contamination in French estuaries. Appl Environ Microbiol 73: 4857–4866.
King EL, Bachoon DS, Gates KW . (2007). Rapid detection of human fecal contamination in estuarine environments by PCR targeting of Bifidobacterium adolescentis. J Microbiol Methods 68: 76–81.
Lee JE, Lim MY, Kim SY, Lee S, Lee H, Oh HM et al. (2009). Molecular characterization of bacteriophages for microbial source tracking in Korea. Appl Environmen Microbiol 75: 7107–7114.
Ley RE, Turnbaugh PJ, Klein S, Gordon JI . (2006). Microbial ecology human gut microbes associated with obesity. Nature 444: 1022–1023.
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS et al. (2008). Evolution of mammals and their gut microbes. Science 320: 1647–1651.
Long SC, Arango P C, Plummer JD . (2005). An optimized enumeration method for sorbitol-fermenting Bifidobacteria in water samples. Can J Microbiol 51: 413–422.
Lozupone C, Knight R . (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71: 8228–8235.
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA et al. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 437: 376–380.
Scott TM, Rose JB, Jenkins TM, Farrah SR, Lukasik J . (2002). Microbial source tracking: current methodology and future directions. Appl Environ Microbiol 68: 5796–5803.
Seurinck S, Defoirdt T, Verstraete W, Siciliano SD . (2005). Detection and quantification of the human-specific HF 183 Bacteroides 16 S rRNA genetic marker with real-time PCR for assessment of human faecal pollution in freshwater. Environ Microbiol 7: 249–259.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504.
Simpson J, Santo Domingo J, Reasoner D . (2002). Microbial source tracking: state of the science. Environ Sci Technol 36: 5279–5288.
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE et al. (2009). A core gut microbiome in obese and lean twins. Nature 457: 480–484.
Acknowledgements
This research was supported by The Eco-Technopia 21 Project (900-20080010) and Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2009-0089452).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Lee, J., Lee, S., Sung, J. et al. Analysis of human and animal fecal microbiota for microbial source tracking. ISME J 5, 362–365 (2011). https://doi.org/10.1038/ismej.2010.120
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2010.120
Keywords
This article is cited by
-
Robust host source tracking building on the divergent and non-stochastic assembly of gut microbiomes in wild and farmed large yellow croaker
Microbiome (2022)
-
Bacterial and eukaryotic microbial communities in urban water systems profiled via Illumina MiSeq platform
3 Biotech (2021)
-
Effect of different seasons (spring vs summer) on the microbiota diversity in the feces of dairy cows
International Journal of Biometeorology (2020)
-
Free-living bacteria and potential bacterial pathogens in sewage treatment plants
Applied Microbiology and Biotechnology (2018)
-
Diversity, abundance, and possible sources of fecal bacteria in the Yangtze River
Applied Microbiology and Biotechnology (2017)
