
Abstract
Deep-sea sediment is one of the most important microbial-driven ecosystems, yet it is not well characterized. Genome sequence analyses of deep-sea sedimentary bacteria would shed light on the understanding of this ecosystem. In this study, the complete genome of deep-sea sedimentary bacterium Pseudoalteromonas sp. SM9913 (SM9913) is described and compared with that of the closely related Antarctic surface sea-water ecotype Pseudoalteromonas haloplanktis TAC125 (TAC125). SM9913 has fewer dioxygenase genes than TAC125, indicating a possible sensitivity to reactive oxygen species. Accordingly, experimental results showed that SM9913 was less tolerant of H2O2 than TAC125. SM9913 has gene clusters related to both polar and lateral flagella biosynthesis. Lateral flagella, which are usually present in deep-sea bacteria and absent in the related surface bacteria, are important for the survival of SM9913 in deep-sea environments. With these two flagellar systems, SM9913 can swim in sea water and swarm on the sediment particle surface, favoring the acquisition of nutrients from particulate organic matter and reflecting the particle-associated alternative lifestyle of SM9913 in the deep sea. A total of 12 genomic islands were identified in the genome of SM9913 that may confer specific features unique to SM9913 and absent from TAC125, such as drug and heavy metal resistance. Many signal transduction genes and a glycogen production operon were also present in the SM9913 genome, which may help SM9913 respond to food pulses and store carbon and energy in a deep-sea environment.
Similar content being viewed by others


Introduction
The deep-sea floor accounts for almost 60% of the Earth's surface (Brunnegarda et al., 2004), most of which is covered with fine-grained sediments (Jørgensen and Boetius, 2007). Deep-sea sediment is a dynamic geo- and biosphere that hosts rich microbial communities (Whitman et al., 1998; Jørgensen and Boetius, 2007). It is estimated that around 13% of total global bacteria live in the upper 10 cm of deep-sea sediments (Turley, 2000; Schippers et al., 2005). In most deep-sea sediments, no light is present, temperatures are close to freezing (–1 °C to 4 °C), the pressure is very high and oxygen concentrations are usually very low compared with surface seawater (Jørgensen and Boetius, 2007). These extreme conditions have affected the sedimentary bacteria at various aspects, forcing bacteria to evolve special features to adapt for the deep-sea environment.
Genomic analysis provides an ideal technique for characterizing special features of deep-sea bacteria. The genomic analyses of deep-sea bacteria, such as Photobacterium profundum SS9 (Vezzi et al., 2005), Shewanella piezotolerans WP3 (Wang et al., 2008) and Alteromonas macleodii ‘deep ecotype’ (Ivars-Martinez et al., 2008), have revealed some features common to many deep-sea bacteria. In general, the genomes of cultured deep-sea bacteria contain more transposable and phage related elements and larger intergenic spacers than that of surface bacteria (Ivars-Martinez et al., 2008). Light-related genes, such as photoreactivation genes, are absent in deep-sea bacteria. Conversely, some genes are better represented in the deep-sea bacteria, such as those that are important for both cold and pressure adaptation, such as membrane unsaturation genes (Lauro et al., 2008). Currently, only a few species of deep-sea bacteria have been subjected to sequence analyses. Considering the high diversity of deep-sea bacteria (Toffin et al., 2004; Sogin et al., 2006; Brown et al., 2009), more genomic sequences of representative bacterial strains must be analyzed to fully characterize the special features required for adaptation to deep-sea sediments.
Pseudoalteromonas is a genus of gamma-proteobacteria that is widespread in the world's oceans, from surface water to deep-sea sediments. Pseudoalteromonas species are usually associated with eukaryotic organisms and produce a large amount of biologically active agents, such as extracellular antibiotics, toxins and polysaccarides (Holmstrom and kjelleberg, 1999). Pseudoalteromonas species are cultivable bacteria found consistently in deep-sea sediments and produce large quantities of extracellular enzymes (Chen et al., 2003; Cui et al., 2008; Zhou et al., 2009), suggesting that they have an important role in the decomposition of particulate organic matter (POM) in deep-sea sediments. Although many Pseudoalteromonas strains have been recovered from sediments, the special features and adaptation mechanisms that allow them to thrive in extreme deep-sea conditions have barely been explored at the genomic level. Pseudoalteromonas sp. SM9913, isolated from deep-sea sediment at a water depth of 1855 meters near the Okinawa Trough, is a psychrophilic strain that produces a large quantity of proteases and exopolysaccharides (EPSs), indicating its function in the degradation of sedimentary particulate organic nitrogen and its potential uses in biotechnology applications (Chen et al., 2003; Qin et al., 2007a). Aside from its adaptability to cold temperatures, the deep-sea-specific features of SM9913 remain largely unknown. Pseudoalteromonas haloplanktis TAC125 which was isolated from the Antarctic coastal sea water and has already been sequenced by the whole genome shotgun method (Médigue et al., 2005) is closely related to SM9913. SM9913 and TAC125 are both psychrophilic strains with similar optimal growth temperatures, and both were isolated from permanently cold environments. Therefore, SM9913 and TAC125 represent different ecotypes of Pseudoalteromonas from cold deep-sea sediment and cold surface water, respectively, and are good models for the comparison of properties unique to bacteria in deep-sea sediment or surface seawater.
Genome sequence can provide a path toward understanding of how bacteria adapt to their environments. For example, the complete genome sequence analysis of P. haloplanktis TAC125 reflects its ability to cope with cold in Antarctic sea water (Médigue et al., 2005). Genome analysis of Pseudoalteromonas tunicata isolated from the surface of marine alga reflects its surface-associated marine lifestyle (Thomas et al., 2008). Comparative genomics is a powerful tool for understanding the ecological specialization of different ecotypes, which may shed light on the specific adaptations that bacteria make in different microenvironments (Ting et al., 2002; Kettler et al., 2007; Ivars-Martinez et al., 2008).
In this study, the complete genome sequence of P. sp. SM9913 is analyzed, and compared with the genome of P. haloplanktis TAC125. The genome sequencing and related functional analyses of SM9913 reveal some special features of Pseudoalteromonas that have allowed it to adapt to deep-sea sediment. This represents the first detailed comparative genomic analysis of Pseudoalteromonas species inhabiting dramatically different environments (deep-sea sediment versus surface seawater).
Materials and methods
Bacterial strains
P. sp. SM9913 (hereafter called SM9913) isolated from deep-sea sediment was cultured at 15 °C, the optimal temperature for SM9913 growth (Chen et al., 2003), using marine Luria–Bertani broth: 10 g peptone, 5 g yeast extract, 1 l artificial seawater, pH 7.5. The cells were harvested by centrifugation at 12 000 g at 4 °C for 10 min. DNA was extracted using a DNA extraction kit (Qiagen, Hilden, Germany). P. haloplanktis TAC125 (hereafter called TAC125) was obtained from the Institute Pasteur Collection (collection number CIP108707).
Genome sequencing
The genome sequence of strain SM9913 was determined using a combination of Sanger sequencing and 454 pyrosequencing. About 100 Mb data were obtained from one 454 GS FLX (Genome Sequencer FLX) sequencing run. The resulting sequences were assembled into 110 large contigs that were oriented by Sanger sequencing reads from paired ends of plasmid and fosmid libraries with average insert sizes of 3–5 and 40 kb, respectively. A total of 11 100 Sanger sequencing reads were used in the genome assembly. Gaps were closed by primer walking and PCR segment sequencing. The phred-phrap-consed package (Gordon et al., 1998) was used for assembly and finishing, and the finished genome was further validated by long PCR of 10 kB genome fragments.
Genome annotation and analysis
The tRNA genes were predicted by tRNAscan-SE (Lowe and Eddy, 1997). The rRNA genes were identified by BLAST search against Rfam (Griffiths-Jones et al., 2003) and rRNA gene sequences from TAC125. The open reading frames were found by GLIMMER 3.0 (Delcher et al., 1999). The predicted open reading frames were annotated by similarity searches against databases of nonredundant protein sequences from the National Center for Biotechnology Information (NCBI), clusters of orthologous groups of proteins (COGs) (Tatusova et al., 2001) and InterPro (Apweiler et al., 2001). The annotation of open reading frames was manually curated with Artemis (Rutherford et al., 2000). Signal peptide prediction was performed by SignalP 3.0 (Bendtsen et al., 2004). Clustered regularly interspaced short palindromic repeats (CRISPRs) were found with CRISPR-finder (http://crispr.u-psud.fr/Server/CRISPRfinder.php). The codon adaptation index was calculated by the EMBOSS cai program using all ribosomal protein genes as highly expressed reference genes (Hjerde et al., 2008).
Comparative genomics
The genome sequence of TAC125 was downloaded from NCBI. An all-versus-all search was performed using all proteins of SM9913 and TAC125 by BLASTP with an E-value cutoff of 1e-5. Orthologous proteins are defined as reciprocal best hit proteins with a minimum 50% identity and 70% of the length of the query protein, calculated by the BLAST algorithm. Proteins without orthologs are considered to be specific proteins. The COG function category was analyzed by searching all predicted proteins against the COG database on the basis of the BLASTP; the final results were put together by custom-made Perl scripts (available from the authors upon request). Average nucleotide identity was calculated according to the method of Konstantinidis and Tiedje (Konstantinidis and Tiedje, 2005) using SM9913 as the query genome. Genomic islands (GIs) were identified by G+C content variation across the genome and dinucleotide bias according to the methods of Karlin (Karlin, 2001), as well as the presence of transposable elements and the genes specific for SM9913. Therefore, the GIs identified contain mainly SM9913-specific genes.
Phenotypic characteristics and comparison
H2O2 resistance was tested in marine Luria–Bertani broth at 15 °C. Cultures of SM9913 and TAC125 were grown to an OD600 of 0.6, and then divided equally into four parts. H2O2 was added to a final concentration of 0, 5, 10 and 15 mM, respectively. The cultures were further incubated at 15 °C, and the OD600 of the cultures was measured at 1 h interval. The experiments were repeated twice. Swimming motility was determined using the hanging drop method (Qin et al., 2007b). Sensitivity to antibiotics was tested using the disc-diffusion method, as described previously (Qin et al., 2007b). The antibiotics contained within one disc were: kanamycin, 30 μg; tetracycline, 30 μg; penicillin, 1 μg; ampicillin, 10 μg; amoxicillin, 10 μg; and chloromycetin, 30 μg. Chitin degradation ability was tested according to Cottrell et al. (2000), that is, by observing cleared zones. Other physiological and biochemical properties were tested using the commercial systems API 20E, API ZYM (both from bioMerieux), following the manufacturer's instructions with some modifications (Qin et al., 2007b). Flagella of cells grown in marine Luria–Bertani broth or on 0.3% agar were observed by transmission electron microscopy (JEM-100CXII). The cells were negatively stained with 2% phosphotungstic acid before observation.
Database accession numbers
The complete genome sequence of strain SM9913 was deposited in GenBank under accession nos. CP001796 and CP001797.
Results and discussion
General features
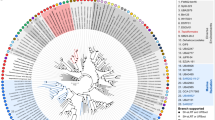
The general features of the SM9913 genome are summarized in Table 1, Figure 1 and Supplementary Table S1. Similar to TAC125, the genome of SM9913 is composed of two chromosomes (chrs), which are 3.3 Mb and 700 kb in size, respectively. The genomes of the two strains are quite similar in structure and size (Supplementary Figure S1). Similar to TAC125, chr II of SM9913 does not show a standard GC skew. This is consistent with the conclusion that these two strains are highly similar (16S rRNA gene identity of both strains is more than 99%, Figure 2). The average nucleotide identity between SM9913 and TAC125 is 85%, which indicates that the two strains are not the same species, but have a high identity, considering their widely divergent dwelling environments. The two chrs of SM9913 contain a total of 3711 predicted coding sequences, 66.9% of which can be annotated with known or predicted functions. The 62 tRNA genes, 8 rRNA operons and 1 extra 5S rRNA gene are all located in chr I. Elongated helices that are found in the 16S rRNA genes of some piezophiles (Lauro et al., 2007) were not detected in SM9913.

Circular representation of the SM9913 genome. From the outside inward: The first and second circles show predicted coding regions transcribed in the clockwise and counterclockwise directions, respectively. Colors indicate COG functional categories: dark grey indicates energy production and conversion (C); red indicates cell-cycle control, cell division and chromosome partitioning (D); green indicates amino-acid transport and metabolism (E); blue indicates nucleotide transport and metabolism (F); cyan indicates carbohydrate transport and metabolism (G); magenta indicates coenzyme transport and metabolism (H); yellow indicates lipid transport and metabolism (I); pale green indicates translation, ribosomal structure and biogenesis (J); light blue indicates transcription (K); orange indicates replication, recombination and repair (L); brown indicates cell wall/membrane/envelope biogenesis (M); pale pink indicates cell motility (N); light grey indicates post-translational modification, protein turnover or chaperones (O); mid red indicates inorganic ion transport and metabolism (P); light red indicates secondary metabolite (Q); pink indicates general function prediction only (R); dark red indicates unknown function (S); purple indicates signal transduction mechanisms (T); gold indicates intracellular trafficking, secretion and vesicular transport (U); navy blue indicates defense mechanisms (V); and black indicates hypothetical proteins. The third and fourth circles show the rRNA and tRNA, respectively. The fifth circle shows GIs. The sixth and seventh circles show percent G+C content and GC skew plot, respectively.

Phylogenetic tree based on the 16S rRNA gene sequences of SM9913 and related strains with completely sequenced genomes. The tree was generated by the neighbor-joining method using MEGA 3.1.
SM9913 appears to possess all the genes of the glycolysis, tri-carboxylic acid cycle and pentose phosphate pathways. This indicates that SM9913 can metabolize glucose to generate energy, nicotinamide adenine dinucleotide and nicotinamide adenine dinucleotide phosphate. The SM9913 genome also contains the key genes of the Entner–Doudoroff pathway, 6-phosphogluconate dehydratase (PSM_A1394) and 2-keto-3-deoxy-6-phosphogluconate aldolase (PSM_A1395). It harbors two sucrose phosphorylase genes (PSM_B0264 and PSM_B0464) and does not contain the genes required to use mannose or galactose. This is consistent with experimental results that SM9913 can use sucrose, but not mannose (Table 2). SM9913 has one glutamine synthetase gene ( PSM_A0181), which is related to ammonium assimilation (Brunnegarda et al., 2004). The strain cannot use nitrite or nitrate, consistent with the finding that it does not possess nitrite reductase or nitrate reductase genes.
SM9913 and TAC125 share 2698 orthologous genes (Figure 3a), accounting for 72.7% and 77.4% of all the genes of SM9913 and TAC125, respectively. There is no significant difference in amino-acid composition between the two strains (data not shown). As shown in Figure 3b, a larger proportion of SM9913-specific genes belonging to COGs representing the N (cell motility) and T (signal transduction mechanisms) groups, and fewer, unique genes in the I (lipid transport and metabolism) and Q (secondary metabolites biosynthesis, transport and catabolism) groups, compared with those of TAC125.

Comparison of the gene content of SM9913 and TAC125. (a) Venn diagram of the orthologous and specific genes in each strain. (b) The COG categories of the orthologous and specific genes in each strain. COG functional categories are described in Figure 1.
A total of 12 GIs larger than 15 kb can be identified by the methods described in the materials and methods section. Eleven GIs are located in chr 1 and one GI is located in chr 2 (Figure 1). General information about these GIs is summarized in Table 3. The average codon adaptation index of the GIs is similar to the average genome composition. Most genes in the GIs of SM9913 are specific genes that do not have orthologs in TAC125. These specific genes may confer some features of SM9913 that differentiate it from TAC125, which will be discussed in detail later.
Signal transduction genes
Histidine kinase and GGDEF domain-containing proteins are more enriched in deep sea water microbes than in surface sea water microbes (Konstantinidis et al., 2009). Histidine kinase is the sensing component of the two-component signal transduction system, and the GGDEF domain-contained protein is involved in cyclic diguanosine monophosphate synthesis (Römling et al., 2005). Cyclic diguanosine monophosphate is a novel global second messenger in bacteria, affecting multicellular behaviors, such as motility, phage and heavy metal resistance, EPS production and biofilm formation (Römling et al., 2005; Römling, 2009). The SM9913 genome contains 27 genes encoding GGDEF domain-containing protein and 5 genes encoding diguanylate cyclase that catalyzes the formation of cyclic diguanosine monophosphate. Meanwhile, TAC125 has only 15 genes encoding GGDEF domain-containing protein and no diguanylate cyclase genes. In addition, SM9913 harbors 33 histidine kinase genes in its genome, whereas TAC125 has only 20. The difference in the sizes of these families is primarily responsible for the fact that SM9913 has more specific genes belonging to signal transduction systems than does TAC125 (Figure 3b).
In the deep-sea bacterium Ph. profundum SS9 more signal transduction genes are overexpressed at 28 mega Pascal than at 0.1 mega Pascal (Vezzi et al., 2005). This shows that signal transduction genes have an important role in the adaptation of bacteria to deep-sea conditions. POM that have reached the sea bed from surface primary production is the major nutrients resource for deep-sea bacteria (Pfannkuche, 1992; Boetius et al., 2000; Smith et al., 2008). Every year, most nutrients arrive at the deep sea in pulse (Smith et al., 2008; Witte et al., 2003a, 2003b). Deep-sea bacteria must respond to this nutrient pulse quickly as well. With its additional signal transduction genes, SM9913 may sense seasonal influx of POM keenly in the deep-sea environment.
Transposable elements
Transposases and integrases can mediate the movement of DNA sequences to new locations within or between genomes (Rice and Baker, 2001). SM9913 contains 28 transposase genes and 12 integrase genes, whereas TAC125 contains only 13 and 7, respectively. Konstantinidis et al. also reported that deep-sea bacteria usually have more transposable elements than bacteria in surface sea water (Konstantinidis et al., 2009). This implies that there are more gene transfer incidents in deep-sea bacteria than in surface-sea bacteria. About one third of the transposase and integrase genes of SM9913 are located in GIs 6, 8, 9 and 11; GI-8 and GI-9 are related to heavy metal resistance (Table 3). The transposases and integrases may have function in the transfer of these resistance-related genes into SM9913. Under the strong selective pressure of the deep-sea environment, some adaptive mutations will be advantageous for bacteria to survive. The large number of transposases and integrases will promote the movement of these adaptive genes between different species, which would contribute to the diversity of deep-sea bacteria.
Sensitivity to reactive oxygen species (ROS)
When comparing the proteins involved in cold/salt adaptation between SM9913 and TAC125 (reported for TAC125 in Médigue et al., 2005), we found that the number of these proteins in the two strains is almost the same, indicating that SM9913 and TAC125 have the similar cold/salt adaptation strategies. However, the number of dioxygenase genes in the two strains is quite different. TAC125 has twelve dioxygenase genes, whereas SM9913 has just four (PSM_A0972, PSM_A1718, PSM_A1738 and PSM_B0404). The large number of dioxygenase genes in the genome is a strategy that TAC125 adopts against the ROS production that occurs due to the increased solubility of oxygen at low temperature. Indeed, TAC125 is remarkably resistant to H2O2 (Médigue et al., 2005). SM9913 also lacks the fatty acid metabolism gene cluster of PSHAa0894 to PSHAa0910 that is related to ROS resistance in TAC125 (Médigue et al., 2005). The smaller number of dioxygenase genes and lack of a ROS resistance-related gene cluster in the genome indicate that SM9913 may have lower H2O2 resistance than does TAC125. This was confirmed experimentally (Figure 4). SM9913 was only able to grow at concentrations of up to 5 mM H2O2, whereas TAC125 grew well even at 10–15 mM H2O2. Genome analysis also shows that SM9913 lacks the molybdopterin metabolism that results in ROS production, implying that SM9913 typically faces low ROS concentrations. Consistent with this finding, the oxygen concentration in sea water at a depth of 1800 m is about one third of that in surface sea water (Brown et al., 2009). Furthermore, the oxygen concentration in deep sea sediment is even lower than that in the surrounding deep-sea water (Glud, 2008). The small number of dioxygenase genes in SM9913 and its sensitivity to ROS reflect its long-term life history in deep-sea sediment with low oxygen concentrations.

Growth curves of SM9913 and TAC125 at different concentrations of H2O2. (a) SM9913. (b) TAC125. The Y axis is logarithmic scale.
Exopolysaccharide biosynthesis
GI-1, -5 and -7 of SM9913 are related to EPS biosynthesis (Table 3). The genes in these GIs form three EPS biosynthesis clusters. Our previous study showed that SM9913 can produce large amounts of highly acetylated EPS (Qin et al., 2007a). These EPS can protect the cold-adapted protease MCP-01 produced by SM9913 from autolysis and can bind many metal ions because of their net negative charge and acidic properties (Qin et al., 2007a). Some deep-sea bacteria with sequenced genomes, such as Idiomarina loihiensis (Hou et al., 2004) and A. macleodii ‘deep ecotype’ (Ivars-Martinez et al., 2008), have EPS biosynthesis genes, showing that the production of EPS may be a common strategy that deep-sea bacteria adopt to endure extreme conditions. EPS can endow bacteria with other advantages as well, such as the ability to adhere to and colonize surfaces, and can speed biochemical interactions, protect the cell and concentrate dissolved organic matter in the marine environment (Nichols et al., 2005; Qin et al., 2007a).
Drug and heavy metal resistance
GI-2 of SM9913 contains a gene encoding an AcrA/E family efflux transporter (PSM_A0563) and a gene encoding an AcrB/AcrD/AcrF family protein (PSM_A0564), both of which are related to drug resistance (Nishino and Yamaguchi, 2001). SM9913 also has 12 multidrug-resistance genes and 4 β-lactamase genes. The percentage of SM9913-specific genes defined as ‘defense mechanisms’ by COG is 3.0%, but these genes are only 1.5% of the genes unique to TAC125. The Okinawa Trough is at the edge of the continental shelf of the East China Sea. Compared with Antarctic sea water, the water and sediments in the Okinawa Trough are likely to contain more materials from the continent, including antibiotics (Dang et al., 2009). As a result of adaptation to their respective environments, SM9913 can resist some antibiotics, such as ampicillin, penicillin and amoxicillin, whereas TAC125 is susceptible to them (Table 2).
There are some heavy metal resistance and efflux genes in GI-8 and GI-9 of SM9913. On the basis of the genome survey, SM9913 is likely to be resistant to cadmium, cobalt, copper, magnesium, mercury and zinc. SM9913 also contains a Hg(II)-responsive transcriptional regulator gene (PSM_A2609) and a MerR family transcriptional regulator gene (PSM_A2628) in GI-9, which may be involved in the regulation of metal resistance. These genes may make SM9913 more resistant to some heavy metals than TAC125. Our experimental results showed that SM9913 is more resistant to zinc than TAC125. With 1 mM zinc acetate in the culture, SM9913 grew much faster than TAC125 (Supplementary Figure S2). Most deep-sea isolates are more resistant to heavy metals than surface bacteria. For example, the A. macleodii ‘deep ecotype’ strain is reported to be significantly more resistant to mercury and zinc than the A. macleodii ‘surface’ ecotype strain ATCC 27126 (Ivars-Martinez et al., 2008). It is still unclear why deep-sea bacteria are more resistant to heavy metals than surface sea bacteria. However, SM9913 may be adapted to high metal concentrations because the negatively charged EPS that is secreted by deep-sea bacteria sometimes can adsorb more cations around the cell than are needed (Nichols et al., 2005).
Flagella and motility
Flagellar motility is very important to allow bacteria to move toward favorable conditions, form biofilms and acquire nutrients. Deep-sea bacteria usually have both polar and lateral flagella for swimming and swarming, respectively (Eloe et al., 2008; Wang et al., 2008). Generally, two independent gene clusters are responsible for the synthesis of the two types of flagellar equipment. However, SM9913 seems to have three gene clusters for flagellum biosynthesis (Supplementary Figure S3). Cluster I, which is responsible for the synthesis of the lateral flagellum (LF), spans 32 883 bp from PSM_A0884 to PSM_A0920. The LF gene cluster is located in GI-4 of SM9913, and is absent in TAC125. In the deep-sea piezophilic bacterium Ph. profundum SS9, the LF gene cluster may be acquired by horizontal transfer, is only expressed under high pressure conditions and is absent in its shallow-water, pressure-sensitive relative Ph. profundum 3TCK (Eloe et al., 2008). The deep-sea sedimentary bacterium S. piezotolerans WP3 contains a LF gene cluster that is upregulated at low temperatures (Wang et al., 2008), whereas the surface-sea bacteria Pseudoalteromonas tunicata D2 and Alteromonadales sp. TW-7 both lack the LF gene cluster (Thomas et al., 2008). Generally, it seems that deep-sea sedimentary bacteria usually have the LF gene cluster, whereas related surface bacteria lack it. Because LF is responsible for bacterial swarming, swarming should be an important movement for bacterial life in deep-sea sediments, but not necessary for surface bacteria. Although SM9913 has the LF gene cluster, expression of the LF could not be observed when the strain grew on 0.3% marine LB agar at atmospheric pressure, suggesting that the LF of SM9913 may be expressed only under high pressure conditions.
Cluster II and cluster III are responsible for synthesis of the polar flagellum (PF) in SM9913. Cluster II spans 32 907 bp from PSM_A2229 to PSM_A2262, and cluster III contains 21 756 bp from PSM_A2278 to PSM_A2299. The PF of SM9913 is visible under transmission electron microscopy (Supplementary Figure S4), which endows SM9913 with the ability to swim in sea water. It is surprising that there are two large gene clusters for the synthesis of PF in SM9913, as TAC125 and other PF-containing bacteria have only one PF gene cluster. However, analysis of the GIs shows that the two PF clusters in SM9913 are separated by GI-7. This indicates that the two PF gene clusters were actually a single PF gene cluster that has been interrupted by the insertion of other genes, which may have been horizontally transferred to SM9913. The PF observed indicates that the PF gene cluster is still active in SM9913 and that its function is not significantly affected by the insertion. The PF and LF motors are usually driven by different ion-motive forces in marine bacteria (Atsumi et al., 1992). In SM9913, the putative proton-driven component LafTU (PSM_A0904 and PSM_A0905), responsible for the power of LF, resides in the LF gene cluster. Four genes, motA (PSM_A0714), motB (PSM_A0714), motX (PSM_A2815) and motY (PSM_A2160), which are related to sodium-driven complexes and are associated with PF rotation (Eloe et al., 2008), are also present in the genome. This confirms the presence of two types of flagellar equipment in SM9913.
With its two flagellar systems, SM9913 can swim in the sea water and swarm on the sediment particle surface, which is advantageous in the acquisition of nutrients. Only particulate materials can reach the deep-sea bed, so a surface-adapted motility system could allow SM9913 to attach to and move on particulate materials. SM9913 can form biofilms on water–solid interfaces (data not shown), which indicates its surface-attachment abilities. Attached bacteria tend to have larger genomes than do free-living bacteria have (Turley, 2000). This is the case for SM9913, as its genome is slightly larger than that of TAC125. SM9913 can produce an efficient protease to degrade particulate organic nitrogen (Chen et al., 2003; Zhao et al., 2008), reflecting its POM degradation ability in the deep sea. The surface-attached lifestyle would facilitate SM9913 to degrade POM. When nutrients are depleted, the strain can then swim through sediment pore fluids or seawater to newly arrived POM by PF.
Other GIs and related features
GI-12 contains three adjacent chitinase genes (PSM_B0252, PSM_B0253 and PSM_B0254) with predicted signal peptides, implying that SM9913 can degrade chitin. However, our experimental results show that this is not the case. SM9913 can grow on enriched sea water agar containing chitin, but does not form clearing zones and can not grow on unenriched sea water agar after 7 days. This may be due to its lack of other essential chitin degradation elements, such as chitoporin. The SM9913 genome contains no annotated genes for chitoporin, chitodextrinase, N-aceytl-glucosaminidase or N-acetyl-hexosaminidase, all of which are important for the degradation of chitin (Hjerde et al., 2008). We used the chitoporin protein sequence from Vibrio furnissii (AAF97616) to search the genome using BLASTP program with the E-value of 1e-2 and found no hits. These chitinase genes located in the GI implies that these genes may be laterally transferred, and so the genome lacks other chitin-degraded genes. This may explain why the strain cannot degrade chitin despite the presence of chitinase genes.
The SM9913 genome has 27 genes encoding nuclease excluding ribonucleases. There are three restriction endonuclease genes in the genome, two of which are located in GI-10. A DNA methylase gene (PSM_A2853) is also present in GI-10, next to a restriction endonuclease gene (PSM_A2854), which may form a restriction modification system to degrade foreign DNA elements (Ivars-Martinez et al., 2008). Phages are abundant in deep sea environments and are much more abundant than bacteria (Suttle, 2005, 2007; Sorek et al., 2008). Many bacteria contain CRISPR sequences that are thought to function as an anti-phage defense system, using an RNA-silencing-like mechanism to prevent phage infection (Barrangou et al., 2007; Sorek et al., 2008). Though there is a CRISPR-associated protein (PSM_A2058) in GI-6, we could not detect a CRISPR feature in SM9913 genome using the CRISPR-finder. Rather, a restriction modification system may be responsible for degrading alien DNA and protecting the strain from viral infection.
GI-2 contains one TonB-dependent siderophore receptor gene (PSM_A0556) that may be responsible for adsorbing iron ions from the environment (Moeck and Coulton, 1998; Ghysels et al., 2005). There are a total of three TonB-dependent siderophore receptor genes in the SM9913 genome. Other genes of the siderophore uptake system, such as TonB2 and the TonB system transport proteins, ExbB2 and ExbD2 (Ghysels et al., 2005), are all present in SM9913. Iron is always a growth-limiting factor for marine bacteria (Qin et al., 2007a). Thus, it is reasonable to assume that the siderophore uptake system helps SM9913 adsorb iron in the marine environment.
Glycogen production
A glycogen production operon, including glucose-1-phosphate adenylyltransferase, glycogen synthase and glycogen branching enzyme, is present in chr II from PSM_B0508 to PSM_B0514. This operon is absent in TAC125 and conserved in Alteromonadales sp. TW-7 and P. tunicata D2 (Thomas et al., 2008). Bacterial glycogen is considered to be a storage product that provides both energy and carbon during starvation periods (Strange, 1968; Preiss, 1984). Nutrients arrive at the deep-sea in pulse seasonally every year (Pfannkuche, 1992; Witte et al., 2003a, 2003b). Therefore, SM9913 must accumulate glycogen with this operon when nutrients are widely available and use the stored glycogen when nutrients are absent from the environment, which would improve its ability to survive in the deep-sea environment.
Conclusion
Genomic and comparative genomic studies reveal some specific features of deep-sea sedimentary bacterium SM9913, such as drug and heavy metal resistance, sensitivity to H2O2 and many signal transduction genes. The predicted particle-associated lifestyle would facilitate SM9913 to use POM and thrive in deep-sea sediment.
Accession codes
References
Apweiler R, Attwood TK, Bairoch A, Bateman A, Birney E, Biswas M et al. (2001). The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res 29: 37–40.
Atsumi T, McCarter L, Imae Y . (1992). Polar and lateral flagellar motors of marine Vibrio are driven by different ion-motive forces. Nature 355: 182–184.
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315: 1709–1712.
Bendtsen JD, Nielsen H, Heijne GV, Brunak S . (2004). Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340: 783–795.
Boetius A, Ferdelman T, Lochte K . (2000). Bacterial activity in sediments of the deep Arabian Sea in relation to vertical flux. Deep-Sea Res. (II Top. Stud. Oceanogr.) 47: 2835–2875.
Brown MV, Philip GK, Bunge JA, Smith MC, Bissett A, Lauro FM et al. (2009). Microbial community structure in the North Pacific ocean. ISME J 3: 1374–1386.
Brunnegarda J, Grandel S, Stahl H, Tengberg A, Hall POJ . (2004). Nitrogen cycling in deep-sea sediments of the Porcupine Abyssal Plain, NE Atlantic. Progress in Oceanography 63: 159–181.
Chen XL, Zhang YZ, Gao PJ, Luan XW . (2003). Two different proteases produced by a deep-sea psychrotrophilc bacterial strain, Pseudoaltermonas sp. SM9913. Mar Biol 143: 989–993.
Cottrell MT, Wood DN, Yu L, Kirchman DL . (2000). Selected chitinase genes in cultured and uncultured marine bacteria in the alpha- and beta-subclasses of the proteobacteria. Appl Environ Microbiol 66: 1195–1201.
Cui Z, Lai Q, Dong C, Shao Z . (2008). Biodiversity of polycyclic aromatic hydrocarbon-degrading bacteria from deep sea sediments of the Middle Atlantic Ridge. Environ Microbiol 10: 2138–2149.
Dang HY, Zhu H, Wang J, Li T . (2009). Extracellular hydrolytic enzyme screening of culturable heterotrophic bacteria from deep-sea sediments of the Southern Okinawa Trough. World J Microb Biot 25: 71–79.
Delcher AL, Harmon D, Kasif S, White O, Salzberg SL . (1999). Improved microbial gene identification with GLIMMER. Nucleic Acids Res 27: 4636–4641.
Eloe EA, Lauro FM, Vogel RF, Bartlett DH . (2008). The deep-sea bacterium Photobacterium profundum SS9 utilizes separate flagellar systems for swimming and swarming under high-pressure conditions. Appl Environ Microbiol 74: 6298–6305.
Ghysels B, Ochsner U, Mollman U, Heinisch L, Vasil M, Cornelis P et al. (2005). The Pseudomonas aeruginosa pirA gene encodes a second receptor for ferrienterobactin and synthetic catecholate analogues. FEMS Microbiol Lett 246: 167–174.
Glud RN . (2008). Oxygen dynamics of marine sediments. Mar Biol Res 4: 243–289.
Gordon D, Abajian C, Green P . (1998). Consed: a graphical tool for sequence finishing. Genome Res 8: 195–202.
Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR . (2003). Rfam: an RNA family database. Nucleic Acids Res 31: 439–441.
Hjerde E, Lorentzen MS, Holden MTG, Seeger K, Paulsen S, Bason N et al. (2008). The genome sequence of the fish pathogen Aliivibrio salmonicida strain LFI1238 shows extensive evidence of gene decay. BMC Genomics 9: 616.
Holmstrom C, kjelleberg S . (1999). Marine Pseudoalteromonas species are associated with higher organisms and produce biologically active extracellular agents. FEMS Microbiol Ecol 30: 285–293.
Hou S, Saw JH, Lee KS, Freitas TA, Belisle C, Kawarabayasi Y et al. (2004). Genome sequence of the deep-sea gamma-proteobacterium Idiomarina loihiensis reveals amino acid fermentation as a source of carbon and energy. Proc Natl Acad Sci USA 101: 18036–18041.
Ivars-Martinez E, Martin-Cuadrado A, Auria GD, Mira A, Ferriera S, Johnson J et al. (2008). Comparative genomics of two ecotypes of the marine planktonic copiotroph Alteromonas macleodii suggests alternative lifestyles associated with different kinds of particulate organic matter. ISME J 2: 1194–1212.
Jørgensen BB, Boetius A . (2007). Feast and famine-microbial life in the deep-sea bed. Nat Rev Microbiol 5: 770–781.
Karlin S . (2001). Detecting anomalous gene clusters and pathogenicity islands in diverse bacterial genomes. Trends Microbiol 9: 335–343.
Kettler GC, Martiny AC, Huang K, Zucker J, Coleman ML, Rodrigue S et al. (2007). Patterns and implications of gene gain and loss in the evolution of prochlorococcus. PLoS Genet 3: e231.
Konstantinidis KT, Braff J, Karl DM, Delong EF . (2009). Comparative metagenomic analysis of a microbial community residing at a depth of 4000 meters at station ALOHA in the north pacific subtropical gyre. Appl Environ Microbiol 75: 5345–5355.
Konstantinidis KT, Tiedje JM . (2005). Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci USA 102: 2567–2572.
Lauro FM, Chastain RA, Blankenship LE, Yayanos AA, Bartlett DH . (2007). The unique 16S rRNA genes of piezophiles reflect both phylogeny and adaptation. Appl Environ Microbiol 73: 838–845.
Lauro FM, Tran K, Vezzi A, Vitulo N, Valle G, Bartlett DH . (2008). Large-scale transposon mutagenesis of Photobacteriumprofundum SS9 reveals new genetic loci important for growth at low temperature and high pressure. J Bacteriol 190: 1699–1709.
Lowe TM, Eddy SR . (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25: 955–964.
Médigue C, Krin E, Pascal G, Barbe V, Bernsel A, Bertin PN et al. (2005). Coping with cold: the genome of the versatile marine Antarctica bacterium Pseudoalteromonas haloplanktis TAC125. Genome Res 15: 1325–1335.
Moeck GS, Coulton JW . (1998). TonB-dependent iron acquisition: mechanisms of siderophore-mediated active transport. Mol Microbiol 28: 675–681.
Nichols CA, Guezennec J, Bowman JP . (2005). Bacterial exopolysaccharides from extreme marine environments with special consideration of the southern ocean, sea ice, and deep-sea hydrothermal vents: a review. Mar Biotechnol 7: 253–271.
Nishino K, Yamaguchi A . (2001). Analysis of a complete library of putative drug transporter genes in Escherichia coli. J Bacteriol 183: 5803–5812.
Pfannkuche O . (1992). Benthic response to the sedimentation of particulate organic matter at the BIOTRANS station, 47°N, 20°W. DS Res II—Deep-Sea Res 40: 135–149.
Preiss J . (1984). Bacterial glycogen synthesis and its regulation. Annu Rev Microbiol 38: 419–458.
Qin GK, Zhu L, Chen XL, Wang PG, Zhang YZ . (2007a). Structural characterization and ecological roles of a novel exopolysaccharide from the deep-sea psychrotolerant bacterium Pseudoalteromonas sp. SM9913. Microbiology 153: 1566–1572.
Qin QL, Zhao DL, Wang J, Chen XL, Dang HY, Li TG et al. (2007b). Wangia profunda gen. nov., sp. nov., a novel marine bacterium of the family Flavobacteriaceae isolated from southern Okinawa Trough deep-sea sediment. FEMS Microbiol Lett 271: 53–58.
Rice PA, Baker TA . (2001). Comparative architecture of transposase and integrase complexes. Nat Struct Biol 8: 302–307.
Römling U . (2009). Rationalizing the evolution of EAL domain-based cyclic di-GMP-specific phosphodiesterases. J Bacteriol 191: 4697–4700.
Römling U, Gomelsky M, Galperin MY . (2005). C-di-GMP: the dawning of a novel bacterial signalling system. Mol Microbiol 57: 629–639.
Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA et al. (2000). Artemis: sequence visualization and annotation. Bioinformatics 16: 944–945.
Schippers A, Neretin LN, Kallmeyer J, Ferdelman TG, Cragg BA, Parkes RJ et al. (2005). Prokaryotic cells of the deep sub-seafloor biosphere identified as living bacteria. Nature 433: 861–864.
Smith CR, De Leo FC, Bernardino AF, Sweetman AK, Arbizu PM . (2008). Abyssal food limitation, ecosystem structure and climate change. Trends Ecol Evol 23: 518–528.
Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR et al. (2006). Microbial diversity in the deep sea and the underexplored ‘rare biosphere’. Proc Natl Acad Sci USA 103: 12115–12120.
Sorek R, Kunin V, Hugenholtz P . (2008). CRISPR—a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat Rev Microbiol 6: 181–186.
Strange RE . (1968). Bacterial ‘glycogen’ and survival. Nature 220: 606–607.
Suttle C . (2005). Viruses in the sea. Nature 437: 356–361.
Suttle C . (2007). Marine viruses—major players in the global ecosystem. Nat Rev Microbiol 5: 801–812.
Tatusova RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS et al. (2001). The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res 29: 22–28.
Ting CS, Rocap G, King J, Chisholm SW . (2002). Cyanobacterial photosynthesis in the oceans: the origins and significance of divergent light-harvesting strategies. Trends Microbiol 10: 134–142.
Thomas T, Evans FF, Schleheck D, Mai-Prochnow A, Burke C, Penesyan A et al. (2008). Analysis of the Pseudoalteromonas tunicata genome reveals properties of a surface-associated life style in the marine environment. PLoS One 3: e3252.
Toffin L, Webster G, Weightman AJ, Fry JC, Prieur D . (2004). Molecular monitoring of culturable bacteria from deep-sea sediment of the Nankai Trough, Leg 190 Ocean Drilling Program. FEMS Microbiol Ecol 48: 357–367.
Turley C . (2000). Bacteria in the cold deep-sea benthic boundary layer and sediment-water interface of the NE Atlantic. FEMS Microbiol Ecol 33: 89–99.
Vezzi A, Campanaro S, D’Angelo M, Simonato F, Vitulo N, Lauro FM et al. (2005). Life at depth: Photobacterium profundum genome sequence and expression analysis. Science 307: 1459–1461.
Wang F, Wang J, Jian H, Zhang B, Li S, Wang F et al. (2008). Environmental adaptation: genomic analysis of the piezotolerant and psychrotolerant deep-sea iron reducing bacterium Shewanella piezotolerans WP3. PLoS One 3: e1937.
Whitman WB, Coleman DC, Wiebe WJ . (1998). Prokaryotes: The unseen majority. Proc Natl Acad Sci USA 95: 6578–6583.
Witte U, Aberle N, Sand M, Wenzhöfer F . (2003a). Rapid response of a deep-sea benthic community to POM enrichment: an in situ experimental study. Mar Ecol Prog Ser 251: 27–36.
Witte U, Wenzhofer F, Sommer S, Boetius A, Heinz P, Aberle N et al. (2003b). In situ experimental evidence of the fate of a phytodetritus pulse at the abyssal sea floor. Nature 424: 763–766.
Zhao GY, Chen XL, Zhao HL, Xie BB, Zhou BC, Zhang YZ . (2008). Hydrolysis of insoluble collagen by deseasin MCP-01 from deep-sea Pseudoalteromonas sp. SM9913: Collagenolytic characters, collagen-binding ability of C-terminal PKD domain and Implication for its novel role in deep-sea sedimentary particulate organic nitrogen degradation. J Biol Chem 283: 36100–36107.
Zhou MY, Chen XL, Zhao HL, Dang HY, Luan WX, Zhang XY et al. (2009). Diversity of both the cultivable protease-producing bacteria and their extracellular proteases in the sediments of the South China Sea. Microb Ecol 58: 582–590.
Acknowledgements
We thank Yi Ren for his help in sequence assembly and data analysis. The work was supported by National Natural Science Foundation of China (30770040), Hi-Tech Research and Development program of China (2007AA091903, 2007AA021306) and COMRA Program (DYXM-115-02-2-6).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Qin, QL., Li, Y., Zhang, YJ. et al. Comparative genomics reveals a deep-sea sediment-adapted life style of Pseudoalteromonas sp. SM9913. ISME J 5, 274–284 (2011). https://doi.org/10.1038/ismej.2010.103
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2010.103
Keywords
This article is cited by
-
New investigation of encoding secondary metabolites gene by genome mining of a marine bacterium, Pseudoalteromonas viridis BBR56
BMC Genomics (2024)
-
Community composition, co-occurrence, and environmental drivers of bacterioplankton community in surface and 50-m water layers in the subarctic North Pacific
Journal of Oceanology and Limnology (2023)
-
A pathway for chitin oxidation in marine bacteria
Nature Communications (2022)
-
Scientific and technological progress in the microbial exploration of the hadal zone
Marine Life Science & Technology (2022)
-
Mono- and multispecies biofilms from a crustose coralline alga induce settlement in the scleractinian coral Leptastrea purpurea
Coral Reefs (2021)
