
Abstract
Soil-borne microbial communities were examined via a functional gene microarray approach across a southern polar latitudinal gradient to gain insight into the environmental factors steering soil N- and C-cycling in terrestrial Antarctic ecosystems. The abundance and diversity of functional gene families were studied for soil-borne microbial communities inhabiting a range of environments from 51°S (cool temperate – Falkland Islands) to 72°S (cold rock desert – Coal Nunatak). The recently designed functional gene array used contains 24 243 oligonucleotide probes and covers >10 000 genes in >150 functional groups involved in nitrogen, carbon, sulfur and phosphorus cycling, metal reduction and resistance and organic contaminant degradation (He et al. 2007). The detected N- and C-cycle genes were significantly different across different sampling locations and vegetation types. A number of significant trends were observed regarding the distribution of key gene families across the environments examined. For example, the relative detection of cellulose degradation genes was correlated with temperature, and microbial C-fixation genes were more present in plots principally lacking vegetation. With respect to the N-cycle, denitrification genes were linked to higher soil temperatures, and N2-fixation genes were linked to plots mainly vegetated by lichens. These microarray-based results were confirmed for a number of gene families using specific real-time PCR, enzymatic assays and process rate measurements. The results presented demonstrate the utility of an integrated functional gene microarray approach in detecting shifts in functional community properties in environmental samples and provide insight into the forces driving important processes of terrestrial Antarctic nutrient cycling.
Similar content being viewed by others


Introduction
Biogeochemical cycles in Antarctic terrestrial habitats are almost exclusively driven by microbial activities. Indeed, due to a principal lack of insect and mammalian herbivores and detritivores at most locations (Smith and Steenkamp, 1992), food webs are primarily driven via bacterial and fungal detritus-based routes (Smith, 1994). Given their relative trophic simplicity, such ecosystems represent useful models for linking primary production and microbial nutrient cycling and disentangling the interrelated processes. Furthermore, some Antarctic ecosystems are being subjected to unprecedented human-induced climate change, the ramifications of which remain mostly unknown.
Although the nitrogen cycle in terrestrial Antarctic is as yet poorly understood, available evidence suggest that nitrogen is the main limiting factor in high altitude and high latitude ecosystems (Mataloni et al., 2000; Shaw and Harte, 2001; Solheim et al., 2004). Thus, even small changes in the nitrogen cycle, as predicted in climate change scenarios (Barnard et al., 2005), are hypothesized to induce disproportionately large shifts in the dynamics of such ecosystems. However, without any recent baseline study across a range of environments, the effects of perturbations on Antarctic soils are hard to predict. In the Antarctic, it was reported that the main source of nitrogen in soils was either from precipitation of volatilized ammonium when in proximity of bird colonies (Christie, 1987) or from N2-fixation by cyanobacteria (Ino and Nakatsubo, 1986). N2-fixation in Antarctica has also been suggested to occur via the action of free-living N2-fixing bacteria and the cyanobionts of lichens (Line, 1992). However, heterotrophic N2-fixation was not reported as a significant source of reduced nitrogen (Pandey et al., 1992), and heterotrophic bacteria are believed to rarely fix N2 in these environments because of energy limitations (Christie, 1987). In the few other studies addressing the N-cycle in Antarctic habitats, Signy Island habitats were reported to be devoid of ammonia-oxidizing bacteria (Vishniac, 1993), and nitrate-respiring bacteria outnumbered denitrifying bacteria, suggesting N conservation in some Antarctic soils (Christie, 1987).
Available evidence suggests that fungi are the dominant decomposers in Antarctic ecosystems, representing a sharp contrast to Arctic systems, where bacteria dominate this function (Walton, 1985). Vegetation type is also thought to play an important role in driving microbial decomposers. In South Georgia (maritime Antarctic), very marked differences in cellulose degradation were observed under different vegetations, and it was concluded that Antarctic decomposition is linked to vegetation type, substratum potential and relative dominance of fungi (Walton, 1985). Freeze–thaw cycles are also believed to play an important role in the carbon cycle in Antarctica, since they change exudation patterns in cryptogams and impose stress upon resident microbes (Tearle, 1987; Melick and Seppelt, 1992; Melick et al., 1994). In addition to differences in litter quantity and quality, the major soluble carbohydrate exudates also differ between higher plants, bryophytes and lichens (Melick and Seppelt, 1992; Melick et al., 1994), providing an additional mechanism by which vegetation type can influence soil-borne metabolic activities.
Although some anecdotal evidence exists regarding C- and N-cycling in Antarctic ecosystems, detailed and systematic data are still mostly lacking. Indeed, the majority of studies addressing polar N- and C-cycles have chiefly involved general process and pool measurements, without regard for the microbial communities responsible for these processes. Realistic in situ process measurement for such remote, low activity, extreme environments are especially difficult to obtain. Within the study presented here, we propose that microarray-based functional gene analysis of the microbial communities responsible for key nutrient cycle functions might offer an avenue to gain insight into the influence of environmental factors on microbial community processes in terrestrial Antarctic habitats.
DNA microarray technologies are rapidly becoming important tools in the analysis of complex microbial communities inhabiting various environments (Wilson et al, 2002; Zhou, 2003). The majority of DNA microarray applications in microbial ecology have focused on determinations of community structure based upon phylogenetic markers such as 16S rRNA genes (Gentry et al., 2006). Although such approaches provide powerful and detailed pictures of microbial community structure in complex environmental samples, they generally provide little insight into microbial functions. In recent years, major efforts have therefore been into the targeting of other functional genes that might provide insight beyond pure phylogenetic characterizations. A robust 50-mer functional gene array platform has been recently demonstrated to be useful in examining multiple functional gene targets, mainly within key gene families involved in microbial nutrient cycling and contaminant degradation (Rhee et al., 2004; Tiquia et al., 2004). With the expansion of available databases and probe design improvements (He et al., 2005; Li et al., 2005), this platform has recently been expanded to allow for the simultaneous detection of over 10 000 gene variants on a single array (He et al., 2007).
The main goal of the present study was to describe the functional aspects of microbial communities involved in soil-borne nutrient cycling across an Antarctic latitudinal gradient. Toward this goal, we used a recently designed functional gene microarray system, the GeoChip (He et al., 2007), containing probes for the known diversity of the most important microbial driven nutrient cycle processes, especially the N- and C-cycles. Functional community analyses focused on soil samples taken from different soil environments at five sites with latitudes ranging from 51°S (cool temperate – Falkland Islands) to 72°S (cold rock desert – Coal Nunatak) and included a comparison of vegetated versus principally bare sites. We provide a detailed analysis of functional community structure across the study transect and attempt to relate functional community data to specific environmental factors present in these unique environments.
Materials and methods
Sampling sites
During the austral summer of 2003–2004, 2 × 2 m plots were established at the following sites (see Figure 1 for a map): Falklands Islands (cool temperate zone; 51°S 59°W), Signy Island (South Orkney Islands, maritime Antarctic; 60°43′S 45°38′W) and Anchorage Island (near Rothera research station, Antarctic Peninsula; 67°34′S 68°08′W). At each location, two types of vegetation were selected for sampling: (1) ‘vegetated’, where dense vegetation cover was present with retention of underlying soil and (2) ‘fellfield’, represented as rocky or gravel terrain with scarce vegetation or cryptogam coverage. For the Falkland Islands, vegetated sites exhibited a dwarf shrub vegetation (Empetrum rubrum) and the fellfield site was rocky with sporadic grasses (Festuca magellanica and Poa annua). For the locations in the (Maritime) Antarctic, vegetated sites were dominated by mosses (Chorisodontium aciphyllum on Signy Island and Sanionia uncinata on Anchorage Island), and fellfield sites contained lichen cover (principally Usnea antarctica). Twelve plots were delineated per location with half of the plots positioned over each vegetation type. The Falkland Islands fellfield vegetation was not large enough to allow for such a design and nine of the 12 plots were therefore placed in the dwarf shrub vegetation. Two additional sites were chosen for sampling, but without delineation of permanent plots. Six frost polygons at two different sites were sampled near the Fossil Bluff forward operating station (71°19′S 68°18′W), and five frost polygons were sampled from Coal Nunatak (72°03′S 68°31′W). Comparisons between vegetated and fellfield plots were not possible for these last two sites, as they do not support extensive vegetation patches.

Map of the Antarctic Peninsula highlighting the locations of study sites.
Environmental data collection
Automated weather stations and precipitation gauges (Ott hydrometrie, Hoofddrop, The Netherlands) were installed at the first three principal study locations. Within established experimental plots, temperature probes (copper/constantan thermocouple wires) were inserted in the plots 5 cm above the ground, at the soil surface and 5 cm below the soil surface. Soil moisture content was measured with a Water Content Reflectometer (CS616, Campbell Scientific, Shepshed, UK) to a depth of 30 cm. Each of these sensors recorded every hour for the duration of the study, and measurements stored using a data logger (CR10X with a storage module of 16Mb from Campbell Scientific). Soil microclimatic data retrieved from the automated weather stations were averaged over the whole year.
Soil samples
For molecular analyses, five 1 cm diameter (from 2–3 cm up to 15 cm deep) cores were sampled from each plot or polygon. They were frozen to −20°C as soon as possible (within 24 h) and maintained at that temperature until further analysis. Material for soil analyses was collected from a 10-cm diameter core taken directly adjacent to the plots to minimize destructive sampling in the long-term plots. Sampling took place 26–28 October 2004 for the Falkland Islands, 2–3 January 2005 for the Signy Island, 18–19 January 2005 for Anchorage Island and 22–23 February 2005 for Coal Nunatak and Fossil Bluff.
Soil biochemical, physical and enzymatic analyses
Soil analyses were performed via established standard protocols (Carter, 1993). Complete soil analyses and detailed soil biological characterization of the sites are available in Yergeau et al. (2007b). The low amount of soil available for enzymatic activity analyses necessitated modifications of these protocols to accommodate relatively small sample sizes. Nitrogen mineralization and potential nitrification were obtained by mixing 1 g of soil (fresh weight) with 5 ml of phosphate buffer (10 mM KH2PO4) supplemented with 5 mM of (NH4)2SO4 (only for nitrification) and by comparing NO3 and NH4 content at the beginning of the experiment with that measured after 2 weeks of incubation at 20°C. Laccase and cellulase activity were measured on soil water extracts following van der Wal et al. (2006), with the following modification: scaled-down protocols were used here again, using only 1 g of soil (fresh weight) that was extracted with 1 ml water.
Nucleic acid extractions
Soil DNA was extracted after mechanical lysis in a CTAB buffer using a phenol–chloroform purification protocol as detailed in Yergeau et al. (2007b). DNA extractions were performed separately for each of the five subsamples taken per experimental plot. Following PCR-DGGE analysis that confirmed low-intraplot variability (Yergeau et al., 2007b), equal volumes of these five extractions were pooled to create the mixed environmental DNA used for further analysis. After this pooling step, these site/vegetation cover combinations were represented either by nine (Falkland vegetated), six (Signy, Anchorage, Fossil Bluff), five (Coal Nunatak) or three (Falkland fellfield) independent samples (‘biological’ replicates). The whole range of samples was used for all subsequent analyses.
Real-time PCR
The primers and thermocycling regimens used to assess different microbial communities are summarized in Table 1. All PCRs were carried out in 25 μl volumes containing 12.5 μl of ABsolute QPCR SYBR green mix (AbGene, Epsom, UK), 2.5 μl of bovine serum albumin (BSA; 4 mg ml−1), 2.5 μl of each primer (10 μ M). Real-time PCR was performed using an on a Rotor-Gene 3000 (Corbett Research, Sydney, Australia) and mixes were made using a CAS-1200 pipetting robot (Corbett Research, Sydney, Australia) to minimize variation caused by pipetting errors. The amplification protocol consisted of an initial denaturation phase (95°C for 15 min) followed by 40 cycles of denaturation (94°C for 1 min), annealing (specified temperature for 1 min) and elongation (72°C for 1 min), and followed by a final elongation phase (72°C for 10 min). Touchdown protocols were starting at the highest annealing temperature and the temperature was lowered by 1°C each cycle until the final target annealing temperature was reached. The integrity of real-time PCR products was confirmed by melting curve analyses, from 55°C to 98°C. Known template standards were made from whole genomes extracted using a PowerSoil DNA Isolation Kit (Mobio, Carlsbad, CA, USA) from pure bacterial isolates (Table 1). Genomic DNA was then quantified on a ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA) and the number of genomes μl−1 was calculated using the molecular weight of the genome of the closest fully sequenced organism deposited in GenBank. To calculate standard curve values, we assumed that only one copy of each gene was present in the bacterial genome of the organisms used for the standard curves. Since the real-time PCR data was only used for correlation analyses and that this statistical method looks at covariation between two datasets, the exact quantification of the number of copies in a sample is not necessary, and the fact that we assumed only one gene copy per genome for the standards does not affect the results. Furthermore, since the quantification was based on gene copy numbers instead of the number of organisms, the fact that multiple copies or genomes are present in some of the target organisms is not important. Using 10-fold increments, the standard concentrations were adjusted from 106 gene copies μl−1 to 101 gene copies μl−1. Most of the samples and all standards were assessed in at least two different runs to confirm the reproducibility of the quantification.
Rolling circle amplification
Samples from Coal Nunatak and Fossil Bluff did not produce consistent hybridization results with the direct application of labeled DNA to the functional gene microarray. To increase signal levels from these samples, rolling circle amplification was carried using the TempliPhi kit (Amersham, Piscataway, NJ, USA) following manufacturer's instructions. Spermidine (0.1 μg μl−1) and single-strand binding protein (0.04 mM) were added to the reaction mix to facilitate amplification. The reactions were incubated at 30°C for 4 h and the enzyme was then inactivated by incubation at 60°C for 10 min. The amplification products were then used for labeling. Rolling circle amplification has been demonstrated to be particularly well adapted for the amplification of low biomass microbial communities before microarray hybridization (Wu et al., 2006).
DNA labelling
DNA was labelled with cystidine-5 (Cy-5) dye. Briefly, approximately 2 μg of genomic DNA or the whole rolling circle amplification products were denatured for 5 min at 99.9°C in a solution containing 0.1 mM spermidime and 1 × random octamer mix (Invitrogen, Carlsbad, CA, USA) and immediately chilled on ice. Following denaturation, the following components were added: 2.5 mM dithiothreitol (DTT), 0.25 mM dATP, dCTP and dGTP, 0.125 mM dTTP, 0.125 mM Cy5-dUTP, 7 ng μl−1 RecA (Amersham) and 80 U of the large Klenow fragment (Large fragment of DNA polymerase I; Invitrogen). Reaction mixtures were then incubated at 37°C for 6 h. Labeled target DNA was purified with a QIAquick PCR kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions, measured on a ND-1000 spectrophotometer and dried in a speed-vac at 45°C for 45 min. Before hybridization, dried, labelled DNA was resuspended in a solution containing 50% formamide, 5 × sodium saline citrate (SSC) (1 × SSC contained 150 mM NaCl and 15 mM trisodium citrate), 0.1% sodium dodecyl sulfate (SDS), 0.1 μg μl−1 salmon sperm DNA and 0.02 mM spermidine. This solution was incubated at 95°C for 5 min, and after cooling to 60°C, 20 ng μl−1 of RecA protein was added. Labelling reactions were kept at 60°C until the time of hybridization (<30 min).
Microarray hybridization
The GeoChip (He et al., 2007) was used to detect and quantify functional genes in soil in this study. Summarized information about the number of spots and targeted genes in each category of genes present on the GeoChip can be found in Table 2. Hybridizations were performed using a HS4800 Hybridization Station (TECAN US, Durham, NC, USA). The hybridization protocol was as follows. The first wash was carried at 50°C for 1 min with a prehybridization solution (5 × SSC, 0.1% SDS and 0.1% BSA) followed by a 45-min prehybridization. The slides were then washed four times with water at 23°C for 5 min with 30 s soaking. Labelled DNA dissolved in the hybridization solution was then injected in the chamber at 60°C and hybridization was carried at 50°C for 4 h with high agitation. Slides were then washed four times at 50°C for 1 min with a 1.5 min soaking using wash buffer I (1 × SSC, 0.1% SDS), four times at 23°C for 1 min with a 1.5 min soaking using wash buffer II (0.1 × SSC, 0.1% SDS) and four times at 23°C for 1 min with a 2 min soaking using wash buffer III (0.1 × SSC). Slides were finally dried at 23°C for 3 min under a flow of nitrogen gas.
Scanning and image processing
Microarrays were scanned using a ProScanArray microarray scanner (PerkinElmer, Boston, MA, USA), and the associated software, initially at a resolution of 30 μm to obtain a quick display image and then at 10 μm for data collection. The emitted fluorescent signal was detected by a photomultiplier tube (PMT) at 633 nm using a laser power of 95% and a PMT gain of 80%. Images were then transferred to ImaGene 6.0 (BioDiscovery, El Segundo, CA, USA), where a grid of individual circles defining the location of each DNA spot on the array was superimposed on the image to designate each fluorescent spot to be quantified. The raw data were exported to Excel for further data processing.
Owing to the highly variable total hybridization signal between the different samples examined, as well as wide ranges in spot intensities, we found that spot calling based on signal to noise ratios, as typically performed for whole-genome microarray experiments (Verdnick et al., 2002), were not a reliable means of calling spots. Based upon comparison to 16S rRNA gene control spots, we found that the sample with the fewest number of reliable positive spots was in vegetated plots on Signy Island, with approximately 250 positive signals. To allow comparison across microarray hybridizations of different sites, we chose this cutoff for all samples, and scored only the 250 most intense spots of any array as positive. This analysis method is thought to enable robust comparison between samples from widely different environments, but reduces power to compare total diversity and functional redundancy. Although some clearly visible spots may be scored as negative for some samples using our conservative scoring approach, these genes would be expected to be at relatively low levels and therefore presumably less important to the functioning of the systems in question.
The microarray design included one, two or three (and in a few case more) probes for each gene sequence or each group of homologous sequences. Since the multiple probes of a single gene were designed as not targeting exactly the same region of the gene (thus, probe specificities and signals varied widely), a gene was considered as present as soon as one of its probes was scored as present. In most cases where not all probes for the same target were detected, it was the probe that was fitting most closely to the probe design criteria that was scored among the 250 most intense signals. Genes whose probes were not included among the top 250 scored within a sample were scored as below detection limits, yielding a binary presence/absence matrix that was used in subsequent statistical analyses. For some analyses, presence/absence data was summed per gene family.
Total abundance of each spot scored as present was simply the intensity of the spot on the microarray. Although these data were used for some analyses, relative intensity (abundance) values for each hybridization signal were calculated to allow comparison across experimental samples. This value was calculated by dividing the intensity of the spots by the sum of the intensity for all the spots scored as present. When, for a particular gene, more than one probe was scored as present, the relative intensity value was averaged over these probes. For some analyses, the relative intensity was summed per gene family.
Statistical analyses
The matrices resulting from image analyses were considered in statistical analyses as ‘species’ abundance or presence/absence matrices. In most analyses we used either the relative intensity or the presence/absence of each gene (10 337 values for each sample) as defined above.
The effects of the location, vegetation cover and their combined interaction on the functional community structure, as observed in microarray relative abundance data, were tested by distance-based redundancy analyses (db-RDA, Legendre and Anderson, 1999). Bray–Curtis coefficients of similarity were first calculated between samples and used for the computing of principal coordinates in the R package (Casgrain and Legendre, 2001). When necessary, eigenvectors were corrected for negative eigenvalues using the procedure of Lingoes (1971) and then exported to Canoco (version 4.5 for Windows, ter Braak and Šmilauer, 2002) as ‘species data’, where RDA were performed. To test the effects of each of the two variables (vegetation cover and location), they were recoded using dummy binary variables and used in Canoco as the only environmental variable in the model while the other variable was enter as a covariable. To test the interaction, the only variable entered in the model was the interaction between location and vegetation cover while both of them were included (without interaction) as covariables. The significances of these models were tested with 999 permutations. The trace (sum of all canonical eigenvalues) of the models divided by the trace of unconstrained analysis (PCA) gave the percentage of the variation due to the factor tested (Borcard et al., 1992).
To test the influences of environmental and soil variables on the microbial population structure, canonical correspondence analyses (CCA) were carried in Canoco. Relative gene abundance was used as ‘species’ data while soil and environmental data were included in the analysis as ‘environmental’ variables. Rare ‘species’ (in our case, genes) were taken out of the analyses following an empirical method described by D Borcard (http://biol10.biol.umontreal.ca/BIO6077/outliers.html). Variables to be included in the model were chosen by forward selection at a P<0.10 baseline. Using only the selected variables, significance of the whole canonical model was tested with 999 permutations.
All analysis of variances (ANOVAs) and correlations analyses were carried out in Statistica 7.0 (StatSoft Inc., Tulsa, OK). For ANOVA, data normality was tested with a Shapiro–Wilks test and variance homogeneity by Levene's test. When data failed to satisfy one of these tests, an appropriate transformation was applied (log, square root or, in a few extreme cases, Box–Cox transformation). Tukey's honestly significant difference (HSD) method modified for unequal sample sizes (Unequal N HSD in Statistica) was used for post hoc comparison with a P=0.05 grouping baseline. Association strengths between different parameters were tested for significant Pearson's linear correlation (r) and also for nonparametric Spearman's correlation (rs). Correlations were considered significant at a P<0.05 baseline and considered to show a strong trend at a P<0.10 baseline.
Results
Direct labelling of genomic DNA extracted from Antarctic soil samples allowed the detection of diverse functional genes on a microarray platform. However, some samples with lower biomass (Fossil Bluff and Coal Nunatak, see Yergeau et al., 2007b) did not yield consistent hybridization values upon direct analysis, and a linear rolling circle amplification step was introduced to obtain sufficient material for reliable hybridization. This amplification was demonstrated to be rather unbiased in comparisons of amplified versus direct hybridization experiments (Wu et al., 2006). A wide range of different genes (approximately one-third of all the genes on the array) were detected among the 250 most intense spots scored for the different samples (Table 2). Since the Antarctic environment samples studied here are relatively pristine, genes involved in heavy metal and organic pollutant contamination were not examined in detail, and our analyses were focused primarily on genes involved in the N- and C-cycles, including C-1 metabolism.
Functional community structure
Using db-RDA, a multivariate nonparametric test of hypotheses was produced (Table 2) to test the effect of sampling site and vegetation cover on the distribution of relative gene abundances, as determined by relative abundance microarray hybridization data. These tests were also performed using only binary data (presence–absence of a spot), yielding similar results (data not shown). For the purposes of the tests presented in Table 2, Fossil Bluff and Coal Nunatak samples were excluded since no vegetated versus fellfield comparisons could be made and only two replicate samples per location were examined. However, in a separate comparison of location effects across all of the fellfield sites, including this time Fossil Bluff and Coal Nunatak, results were again similar to those represented in Table 2 (data not shown). In the overall data (‘All’ column, Table 2), approximately 10% of the variation observed in the relative abundance data was explained by the location effect and this was highly significant (P<0.01). Vegetation cover accounted for 4.9% of the variation in the whole microarray data and this was also significant (P<0.05). Although vegetation and location had highly significant effects, they only explained a small amount of the total variation in the dataset, which is to be expected when such a small number of variables are entered into such a model (Borcard et al., 1992). These effects were also observed when separately analyzing only the genes associated with the N- and C-cycles. Heavy metal- and organic compound-related genes also showed similar effects of location and vegetation cover. For S-cycle-related genes, there were no significant effects observed for these two variables. Methane-cycle genes revealed a trend (P<0.10) with respect to the influence of location. This location effect was highly significant (P<0.01) when the FB and CN sites we included in the analysis, indicating that these two sites were showing highly different CH4-related genes.
CCA were performed to identify the environmental factors (presented in Table 4) that had the most significant influence on the functional community structure (patterns of relative abundance of the different genes involved in a particular cycle). For the N-cycle-related data subset, the environmental factors that yielded P<0.10 after forward selection were: pH (P=0.07), number of freeze–thaw cycles (P=0.019) and C:N ratio (P=0.095). Together, these factors were forming a significant model (P=0.0010) explaining the patterns in the N-cycle-related genes detected on the array. For the C-cycle-related genes, the most significant environmental factors were: pH (P=0.062), number of freeze–thaw cycles (P=0.005), C:N ratio (P=0.034), NH4+ (P=0.001) and mean soil temperature (P=0.056). Together these factors were also forming a significant model (P=0.0010). For the CH4-cycle genes, the environmental factors retained by forward selection were pH (P=0.025) and number of freeze–thaw cycles (P=0.048), and together they formed a significant model (P=0.0020).
Most highly detected N- and C-cycles genes
Different key gene functions, for both the N- and C-cycles, were represented among those with the highest relative abundance across the sampling sites examined (Supplementary Table S1). N2-fixation genes (nifH) were found among the most strongly detected genes only in bare plots, whereas Signy plots possessed relatively high levels of denitrifying genes (nirK and narG). For Anchorage fellfield plots and Fossil Bluff frost polygons, the functional genes showing the highest levels of detection were putatively coming from members of the Firmicutes. The Fossil Bluff site also contained high levels of Archaeal functional genes, and the three most highly detected genes (a Cyanobacterial urease, an Euryarchaeal gdh and a Firmicutes nifH, see Supplementary Table S1) were unique to this site among the highly detected genes. The only samples that did not have any genes related to nitrification, nitrogen fixation and mineralization among the most highly detected genes were the vegetated plots on Anchorage Island. Interestingly, although urease genes were detected in several different sites among the dominant genes, genes from different organisms were detected in each case, suggesting that different organisms were primarily responsible for this function in the different study sites.
For the C-cycle, genes related to mannase were only detected at high levels in fellfield sites from Signy, Fossil Bluff and Coal Nunatak and were not found among the most highly detected genes in any of the vegetated plots. Genes that can be related to fungi were also more often present among the most highly detected genes as latitude increased. Bacterial chitinase genes were also highly detected in southern plots starting from Anchorage Island, although these genes were also strongly detected for the vegetated plots at the Falkland Islands. Inversely, cellulase genes were less often present among most highly detected genes at higher latitude locations.
Number of gene variants
Using the binary presence–absence data, we investigated the number of different gene variants detected within gene families for functions in the N, C and CH4 cycles, to gain insight into possible functional redundancy among the dominant community members. Here again, FB and CN were left out of the analyses because of low number of replicates and less complete sampling design. Among the 16 well-represented gene families pertaining to the C- and N-cycles {N2-fixation (nif), ammonia oxidation (amo), glutamate dehydrogenase (gdh), urease, assimilatory nitrate reductase (nasA), nitrate reductase (nar), nitrous oxide reductase (nosZ), nitrite reductase (nir), nitric oxide reductase (norB), methane generation, methane oxidation, cellulase, laccase, chitinase, mannase, C-fixation}, six showed a response to either location, vegetation or their interaction term when tested by ANOVA (amo, gdh, urease, methane generation, methane oxidation, C-fixation; Figure 2 and Table 3). Genes related to mineralization (urease), immobilization (gdh) and nitrification (amo) were all influenced only by the interaction term. This is visible in Figure 2 as the differences between vegetated and fellfield plots are changing depending on the site. The number of genes related to methane oxidation was also following a similar pattern, but at a greater level of significance (Table 3). The number of gene variants involved in C-fixation was only influenced significantly by the vegetation cover (Figure 2). The number of genes involved in C-fixation was always higher in fellfield plots and relatively constant across sampling locations. With respect to the number of gene variants related to methane generation, there was a trend towards a decrease with increasing latitude along the main sampling sites, but Fossil Bluff and Coal Nunatak do not follow this trend, displaying high numbers of these gene variants.

Number of gene variants among the 250 most abundant spots belonging to different gene families that were influenced either by location, vegetation cover or location* vegetation cover at the Falkland Islands, Signy Island, Anchorage Island, Fossil Bluff and Coal Nunatak. ▪: vegetated plots; □: fellfield plots. The value represented is the average of the different biological replicates and errors bars are representing the standard error. See Table 3 for ANOVA results.
Relative abundance of different gene families
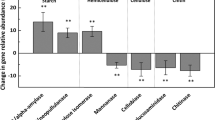
Absolute array hybridization intensities across the study transect mirrored bacterial and fungal biomass estimates as determined by 16S rRNA- and 18S rRNA-specific real-time PCR (Yergeau et al., 2007b). To avoid biases due to these differences in total signal intensity, the relative intensity for each gene family was summed and compared across the different environments using ANOVA (again excluding Fossil Bluff and Coal Nunatak). The seven gene families that showed significant trends are presented in Figure 3 with associated ANOVA tests in Table 3. The relative abundance of genes related to N2-fixation was slightly different for each location, being lower on Signy Island. Glutamate dehydrogenase (gdh) and methane oxidation genes varied in a similar way in response to location and vegetation cover. Vegetated plots harbored relatively more of these gene targets on Falkland and Signy Islands, with the reverse observed for Anchorage Island. Both gene families were also relatively abundant in Fossil Bluff with intermediate levels observed for Coal Nunatak. Nitrite reductase (nir) and cellulases genes showed the same trend, generally decreasing in relative abundance with increasing latitude, while the differences between vegetated and bare plots depended on the location. Nitrate reductase (nar) and C-fixation genes were influenced only by the presence of vegetation, but in opposite directions: nitrate reductase (nar) genes were relatively more abundant in vegetated plots, and C-fixation genes in fellfield plots.

Relative abundance of genes belonging to different gene families that were influenced either by location, vegetation cover or location*vegetation cover at the Falkland Islands, Signy Island, Anchorage Island, Fossil Bluff and Coal Nunatak. ▪: vegetated plots; □: fellfield plots. The value represented is the average of the different biological replicates and errors bars are representing the standard error. See Table 3 for ANOVA results.
Soil variables used for correlation analysis with relative gene abundance are presented in Table 4. The trends in this dataset were already presented and discussed in detail elsewhere (Yergeau et al., 2007b). The correlation of relative gene abundance with different soil and environmental parameters yielded results comparable to the ANOVA tests (Table 5). Genes that appeared to be influenced by location in ANOVA tests were most significantly correlated with variables that covaried with location (NO3−, pH, P, and mean temperature). The genes that were influenced by vegetation cover in ANOVA tests were likewise most correlated with variables that covaried with vegetation cover (organic C, total N, water content and K). Some significant correlations that warrant note include the negative correlations between nitrite reductase (nir), nitric oxide reductase (nor) and methane oxidation gene abundances with NO3− concentration in soil. C-fixation gene abundances were negatively correlated with organic carbon, and cellulose, nitrite reductase (nir) and methane-generating genes were positively correlated with mean soil temperature.
Real-time PCR, enzymatic activities and process rates
Relative gene abundances for nirS, nasA and nifH, as determined using the functional microarray analysis (percentage of total signal), were significantly correlated (r=0.4070, P=0.032, N=28; rs=0.4054, P=0.044, N=25; r=0.5935, P=0.025, N=14; respectively) with the number of gene copies estimated per ng of extracted DNA using real-time PCR assays specific for each of these gene families. In contrast, chiA, narG and nosZ abundances measured by real-time PCR failed to correlate significantly with their respective relative abundances on the microarray.
For selected gene families, enzymatic activities corresponding to a given gene function were determined for comparison with microarray results. Laccase activity in soil extracts showed a highly significant correlation with the total abundance (r=0.6656, P<0.001, N=25) and a strong trend with the relative abundance (r=0.3919, P=0.053, N=25) of this gene family as determined by the microarray. Similar results were observed for cellulase activity, although only when excluding samples showing no measurable activity. Using only the samples that yielded a significant cellulolytic activity, a significant correlation was found between cellulolytic activity and number of different cellulolytic gene variants detected (r=0.7407, P=0.036, N=8) and a strong trend was observed with the total abundance of cellulase genes (r=0.6808, P=0.063, N=8).
Potential process rates measured for two steps of the nitrogen cycle yielded a mixed picture with respect to gene detection via functional microarray experiments. The only functional genes for which absolute microarray detection was correlated with nitrogen mineralization rates were nitrate reductase (nar) genes (r=0.5202, P=0.006, N=26), and a trend was observed with respect to relative abundance of nar genes (r=0.3756, P=0.059, N=26). In contrast, several gene families correlated with potential nitrification rates, including negative correlations with nirK relative abundance (rs=−0.4433, P=0.023, N=26). A similar negative correlation was observed with the summed total of the relative detection of all analysed denitrification genes, (rs=−0.4490, P=0.021, N=26). Also, the relative abundance of glutamate dehydrogenase (gdh) showed a negative trend with respect to nitrification rate (rs=−0.3348, P=0.095, N=26).
Discussion
Ecological application of a functional gene array
This report presents one of the first ecological applications of an expanded functional gene microarray (the GeoChip, He et al., 2007). Although this functional array platform has been shown to provide robust results under controlled experimental conditions (He et al., 2007), application to complex environmental samples across an ecologically relevant range of habitats has yet to be demonstrated. Furthermore, this study represents the first in-depth functional analysis of microbial communities in terrestrial Antarctic ecosystems. The tests of significance performed on microarray patterns across the study sites showed significant effects of location and vegetation cover on C-cycle-related gene distribution (Table 2). N-cycle-related gene distribution was also significantly effected by location and showed a strong trend with respect to vegetation effects. A trend was also observed between location and the distribution of genes related to CH4 transformations. These general results are in good accordance with a previous study that demonstrated that bacterial and fungal community structure, as assessed using PCR-DGGE of rRNA gene fragments, was significantly influenced by location and the vegetation cover (Yergeau et al., 2007b).
Owing to the novelty of this functional gene microarray approach, we sought to confirm detected gene distribution patterns by independent assays, such as quantitative PCR or enzymatic activity assays. In the majority of cases, significant correlations were found between real-time PCR results and the relative gene abundance detected by the array, suggesting a quantitative relationship between microarray signals and environmental gene densities. It should be stressed that the real-time PCR assays relied on different priming sites that those used for probing on the array. Furthermore, probe signals were a summation of signals derived from multiple specific signals within a gene family, whereas real-time PCR results were generated by gene-family specific primers. The relatively low correlation coefficient observed between the real-time PCR and the microarray data can be due to several factors. One reason is that both methods are subject to experimental errors, and that even when using probes and primers targeting the exact same site, correlations between real-time PCR and microarray intensity are not perfect (for example, r=0.87 in Rhee et al., 2004). Another reason is that both methods obviously rely on extant sequence data for primer and probe design and may either miss some members of the target gene families (incomplete coverage) or overlap with related gene families (incomplete specificity). As public databases continue to expand with new microbial functional gene data and improved gene annotation, our ability to increase primer and probe specificity and comprehensiveness should also increase.
The significant correlations between the enzymatic activities measured in soil and the microarray data provide some indication that the detected genes are also expressed in the soil systems examined. Ideally, we would like to examine mRNA levels for the gene functions represented on the functional microarray and efforts are currently underway to realize such a meta-transcriptomics approach (Gao et al., 2007). However, even without such mRNA-based data, it appears that gene density levels offer some predictive value with respect to estimating enzymatic activities in soil ecosystems (at least for cellulolytic and lignolytic activities). Some activity measurements can be coupled directly to the action of a single or very few gene families, making correlations between activity measurements and gene abundances rather straightforward. However, many soil-borne microbial processes, such as mineralization, nitrification and denitrification involve numerous enzymatic steps involving several interrelated microbial populations. For such complex functions, it remains a challenge to understand the metabolic networks involved, and simple process/gene abundance relationships may be less clear. For example, mineralization rates were correlated to nitrate reductase gene detection levels, although this gene family would only be predicted to have an indirect effect on this process. Mineralization is a complex process that is not fully understood, and several of the gene families involved may still be poorly represented on the functional gene array we employed. In general, nitrification is favoured by aerobic conditions while anaerobic conditions favour denitrification, and gene families involved in this later process were inversely related to nitrification rates in our study. In further analyses, we also observed a negative correlation between nitrification rates and methane oxidation genes, which is also a process that is favoured by low oxygen conditions. It is interesting to note that although ammonia monooxygenase (amo) and hydroxylamine oxygenase (hao) catalyze sequential reactions in the ammonia oxidation process, we found no significant correlation in the level of detection of these two gene families. This discrepancy may be to the unequal copy number of these genes in different ammonia-oxidizing bacteria (Hommes et al., 2001) or a different level of probe coverage for these gene families on the array (hao genes were only represented by a few probes). It should also be noted that the functional microarray used in this study does not encompass probes for two other potentially important ammonia oxidation activities, namely anaerobic ammonia oxidation (Strous et al., 1999) and archaeal ammonia oxidation (Leininger et al., 2006).
N-cycle
N2-fixation
Although nitrogen is thought to be the limiting factor in many terrestrial Antarctic ecosystems, extremely little is known about the organisms and genes involved. The functional microarray platform allowed the inspection of Antarctic N-cycle genes in an unprecedented level of detail. N2-fixing genes were most strongly detected in fellfield plots, and the relative abundance was also higher in fellfield plots from the most southern locations, a result that was supported by real-time PCR quantification. However, we observed no evidence of greater N concentration in fellfield sites, and the relative detection level of N2-fixing genes was not related to NO3− or NH4+. This lack of correlation is not entirely unexpected, since gene presence is not necessarily related to activity in soil. Trends in N2-fixing genes may be associated with lichen distribution, as Antarctic lichens have been reported to fix nitrogen via the activities of their bacterial cyanobionts (Line, 1992; Adams et al., 2001). This hypothesis is supported by the fact that N2-fixation, an exclusively prokaryotic trait (Martinez-Romero, 2001), was positively correlated with fungal abundance, an indicator of lichen presence in such systems. Although previous culture-based studies reported cyanobacteria to be the major N2-fixing organisms in the Antarctic (Ino and Nakatsubo, 1986; Line, 1992; Pandey et al., 1992; Vishniac, 1993), this group was only found in low abundance in 16S rRNA gene libraries from these same soils (Yergeau et al., 2007a). Among a range of different Antarctic soils previously studied, the highest rates of N2-fixation were reported for nunatak ‘soils’ (Pandey et al., 1992) and the relatively high abundance of nifH genes at Coal Nunatak follows this pattern. Functional microarray results were in agreement with previous PCR-DGGE analyses specific for 16S rRNA genes of cyanobacteria, plastids and cyanobionts, in which community structure was found to be significantly different across Antarctic study sites and vegetation types (Yergeau et al., 2007b).
Denitrification
For all samples, the genetic potential for complete denitrification (from NO3− to N2) was detected. Interestingly, two of these key gene families (nitrite reductase, nir and nitric oxide reductase, nor) were negatively correlated with soil NO3− concentration (Table 5). Nitrite reductase (nir) catalyzes the first committed denitrification step that leads to a gaseous intermediate (Zumft, 1997). The level of detection of nitrite reductase genes may be related to the low NO3− concentrations in some of the soils examined. For instance, nitrite and nitrate reductase genes were highly detected in Signy Island plots, and these plots were also among the plots with the lowest NO3− concentrations. The relative abundance of nitrite reductase genes, as detected via the functional gene microarray, was also positively correlated with soil temperature and decreased with latitude for the main sampling sites (Figure 3, confirmed by real-time PCR). This temperature dependence may be partially responsible for the relatively high concentrations of NO3− observed on Anchorage Island.
Mineralization, immobilization and nitrification
Urease and ammonia monooxygenase genes were detected in all the soils examined, whereas glutamate dehydrogenase and assimilatory nitrate reductase genes were below the set threshold of detection in several samples. However, the lack of detection of the latter two gene families may simply be due to their relatively poor coverage in public gene databases and, thus, on the functional gene array. The absence of significant correlations between urease gene detection and environmental parameters is not unexpected given the widespread distribution of this trait across microbial groups and habitats. Previous studies have shown that specific groups of ammonia-oxidizing bacteria, and their corresponding ammonia monooxygenase (amo) gene variants, are often associated with key environmental parameters (Kowalchuk and Stephen, 2001), but such distribution patterns were not evident across the transect examined in the present study. However, the number of ammonia monoxygenase (amo) gene variants was highly variable across the study sites (Figure 2), and the dominant gene variant was also different between environments. These results suggest that different ammonia-oxidising bacteria may occur at the different sites, and refutes previous assertions that Antarctic soils are devoid of this functional group (Vishniac, 1993). Again, it is important to keep in mind that genes from archeael ammonia oxidizers (Leininger et al., 2006) have yet to be incorporated into the functional array utilized here.
C-cycle
C-cycle-associated gene distribution was significantly affected by both vegetation cover and location (Table 2). The effect of vegetation cover was probably due to differential input of carbohydrates to soil via exudation, ‘root’ turnover and litter. Importantly, the major soluble carbohydrates in vascular plants, bryophytes and lichens are different (Melick and Seppelt, 1992; Roser et al., 1992; Melick et al., 1994). Thus, the vegetation type is important for providing different quantity and quality of substrates in Antarctic soils. This may be a more important determinant of decomposer community structure than the differences between vascular plant species found in temperate counterparts, since Antarctic systems generally lack aboveground herbivores, thereby channeling a greater proportion of the entire C fixed by the vegetation to the soil. The quantity and quality of C inputs into soil can also be affected by environmental conditions. For instance, the photosynthetic activity of mosses was shown to recover more slowly from cold periods when compared to lichens (Schlensog et al., 2004). Freeze–thaw cycles are also believed to play an important role in C cycling in the Antarctic, not only due to the stress imposed on microbial communities, but also because they induce changes in exudation patterns of cryptogams (Tearle, 1987; Melick and Seppelt, 1992; Melick et al., 1994). It was estimated that each year freeze–thaw induced the release of >15% of the total organic matter of Antarctic cryptogams to the soil microbiota (Tearle, 1987), while only 1.5% of plant material become available each year through the breakdown of dead subsurface material (Davis, 1986). Accordingly, the frequency of freeze–thaw cycles was identified in canonical analyses as a potential driving factor for the structure of the microbes involved in the C-cycle.
C-degradation
Decomposition in Antarctica soils is carried mainly by bacteria, micro-fungi, yeast and probably to some extent by basidiomycetes when present (Smith, 1994). It is also believed that fungi are the dominant decomposers of Antarctica, in contrast to the Arctic where bacteria are presumed to dominate this process (Walton, 1985). Our functional microarray experiments support this hypothesis, as fungal decomposition genes were among the most highly detected genes only in the plots on the Antarctic peninsula and in the bare plots on Signy Island.
We detected fewer cellulase genes among the most dominant genes with increasing latitude, and the total relative abundance of cellulase genes decreased with increasing latitude (Figure 3 and Table 3). The only significant direct correlation with relative cellulase abundance was soil temperature. Temperature has previously been identified as a key determinant of cellulase activity and decomposition rates, particularly in nutrient poor regions such as Antarctica (Pugh and Allsopp, 1982; Kerry, 1990). We also observed that the cellulase gene relative abundance was influenced by vegetation cover in a location-dependent manner, with the Falkland Island plots, which contained vascular plants, being most dissimilar. Similarly, marked differences were previously observed in comparisons of cellulase degradation under different vegetation types on the maritime Antarctic Island of South Georgia (Walton, 1985).
A surprising observation was that laccase genes were more present among dominant genes in plots where no vascular plants (hence true lignin) were present, and this unexpected result was also confirmed by enzymatic assays. This apparent paradox may be explained by the wide specificity of some laccases, which enables them to catalyze a broad range of phenolic substrates other than lignin (Thurston, 1994; Mayer and Staples, 2002). Indeed, lichen, mosses and various microorganisms are known to contain a range of phenolic compounds (Reddy, 1984; Lawrey, 1995), including pigments related to ultraviolet protection (Quesada et al., 1999; Rozema et al., 2001), potentially serving as substrates for fungal laccases (Thurston, 1994; Mayer and Staples, 2002). It was also recently reported that some lichenized ascomycetes showed extracellular laccase activity (Laufer et al., 2006), which could partly explain the results presented here.
C-fixation
Microbial C-fixation genes were always significantly more abundant in fellfield plots (Figure 3). These plots were the ones with the lowest organic carbon and C-fixation gene abundance was negatively correlated with total organic carbon (Table 5). In such environments, microbial C-fixation could be an important input of C to the soil (Vincent, 1988; Hopkins et al., 2006). Davis (1981) showed that the main factors influencing organic matter accumulation in Antarctic soils were temperature and organic matter quality (which depended upon vegetation cover). Similarly, we found potential cellulolytic activity (decomposition) to be dependent upon temperature and microbial C-fixation (primary production) dependent upon vegetation cover.
CH4-cycle
The relative abundance of genes associated with methane generation was temperature dependent and decreased with increasing latitude, except at Fossil Bluff and Coal Nunatak (Figure 2). These last two sampling sites may represent unusual cases (Yergeau et al., 2007b), where CH4-cycle-related bacteria are relatively dominant, as observed for 16S rRNA gene clones libraries retrieved from these environments (Yergeau et al., 2007a). In the other soils examined, it appears that organisms with genes related to methane production are relatively less abundant at low temperatures. Methane oxidation, in contrast, was previously reported to be temperature independent (King and Adamsen, 1992), and, similarly, was not correlated to latitude or soil temperature in our study. However, methane oxidation gene relative abundance was significantly and inversely correlated with nitrate concentration in soil. This finding is not surprising given the potential for methanotrophs to influence the N-cycle (Mancinelli, 1995) and inhibition of methane oxidation by nitrite and ammonium (Schnell and King, 1994). An interesting link can be made between methane oxidation and denitrification, since both processes are stimulated by low levels of oxygen in soil (Mancinelli, 1995; Shapleigh, 2000). This might explain the negative relation between methane oxidation gene relative abundance and nitrate concentration in soils. The levels of oxygen in our soils were not measured, and the only rough proxy available is the soil water content, which was not correlated with methane oxidation or denitrification genes. Measurements of methane concentrations in soil would also be valuable to gain further insights into the dynamics of C-1 compound transformations in these soils (Mancinelli et al., 1981).
Conclusions and perspectives
C-, N- and CH4-cycles along a latitudinal transect of Antarctic environment were described here for the first time via functional microarray approach. Results were confirmed using other independent methods such as real-time PCR, enzymatic and process rates measurement. To provide a reliable comparison between the highly divergent sites examined (Yergeau et al., 2007b) and between amplified and nonamplified samples the data, we analyzed a fixed number of clearly visible spots per array, with intensities in the range observed for SSU gene control spots. In that sense, analyses focussed only on data points that fell within the robust dynamic range of the array system used and results presented in this paper are thought to be independent of the starting amount of nucleic acid, potential inhibitors, background nonhybridizing DNA and small variations in hybridization conditions. Obviously, several limitations are yet to be addressed in the use of microarray in environmental studies. The functional gene array used here focused on a selected number of gene families, while other, neglected functions might also be highly important and informative. In addition, the microarray cannot generate information about new sequences types and thus interrogates only the breadth of known diversity within the targeted gene families. Although our results, which targeted DNA showed significant correlations to enzymatic activities in some cases, analysis at the mRNA level would certainly provide greater insight into in situ activities. However, it should be noted that technical challenges and potential biases in methods of mRNA analysis also exist, especially in their application to low activity ecosystems. Analysis of the amount of data generated by microarray technology is also an important bottleneck, but new statistical approaches are showing interesting promise toward the identification of complex patterns and networks (Gentry et al., 2006). The impact of functional gene microarrays could be increased further by attempts to couple such data with detailed (microarray-based) phylogenetic analyses, potentially linking community structure and function.
References
Adams DG, Bergman B, Nierzwicki-Bauer SA, Rai AN, Schüßler A . (2001). Cyanobacterial-plant symbioses. In: Dworkin M (ed). The Prokaryotes: an Evolving Electronic Resource for The Microbiological Community. Springer-Verlag: New York.
Allen AE, Booth MG, Frischer ME, Verity PG, Zehr JP, Zani S . (2001). Diversity and detection of nitrate assimilation genes in marine bacteria. Appl Environ Microbiol 67: 5343–5348.
Barnard R, Leadley PW, Hungate BA . (2005). Global change, nitrification, and denitrification: a review. Glob Biogeochem Cy 19: GB1007.
Borcard D, Legendre P, Drapeau P . (1992). Partialling out the spatial component of ecological variation. Ecology 73: 1045–1055.
Carter MR . (1993). Soil Sampling and Methods of Analysis. CRC Press: Boca Raton, FL.
Casgrain P, Legendre P . (2001). The R package for multivariate and spatial analysis. Montréal, Canada, Département de sciences biologiques, Université de Montréal, version 4.0d6.
Christie P . (1987). Nitrogen in two contrasting Antarctic bryophyte communities. J Ecol 75: 73–94.
Davis RC . (1981). Structure and function of two Antarctic terrestrial moss communities. Ecol Monogr 51: 125–143.
Davis RC . (1986). Environmental-factors influencing decomposition rates in two Antarctic moss communities. Polar Biol 5: 95–103.
Gao HC, Yang ZMK, Gentry TJ, Wu LY, Schadt CW, Zhou JZ . (2007). Microarray-based analysis of microbial community RNAs by whole-community RNA amplification. Appl Environ Microbiol 73: 563–571.
Gentry TJ, Wickham GS, Schadt CW, He Z, Zhou J . (2006). Microarray applications in microbial ecology research. Microb Ecol 52: 159–175.
He ZL, Gentry TJ, Schadt CW, Wu L, Liebich J, Chong SC et al. (2007). GeoChip: a comprehensive microarray for investigating biogeochemical, ecological and environmental processes. ISME J 1: 67–77.
He ZL, Wu LY, Li XY, Fields MW, Zhou JZ . (2005). Empirical establishment of oligonucleotide probe design criteria. Appl Environ Microbiol 71: 3753–3760.
Hommes NG, Sayavedra-Soto LA, Arp DJ . (2001). Transcript analysis of multiple copies of amo (encoding ammonia monooxygenase) and hao (encoding hydroxylamine oxidoreductase) in Nitrosomonas europaea. J Bacteriol 183: 1096–1100.
Hopkins DW, Sparrow AD, Novis PM, Gregorich EG, Elberling B, Greenfield LG . (2006). Controls on the distribution of productivity and organic resources in Antarctic Dry Valley soils. Proc R Soc Lond B Biol Sci 273: 2687–2695.
Ino Y, Nakatsubo T . (1986). Distribution of carbon, nitrogen and phosphorus in a moss community – soil system developed on a cold desert in Antarctica. Ecol Res 1: 59–69.
Kerry E . (1990). Effects of temperature on growth-rates of fungi from sub-Antarctic Macquarie Island and Casey, Antarctica. Polar Biol 10: 293–299.
King GM, Adamsen APS . (1992). Effects of temperature on methane consumption in a forest soil and in pure cultures of the methanotroph Methylomonas rubra. Appl Environ Microbiol 58: 2758–2763.
Kowalchuk GA, Stephen JR . (2001). Ammonia-oxidizing bacteria: a model for molecular microbial ecology. Annu Rev Microbiol 55: 485–529.
Laufer Z, Beckett RP, Minibayeva FV, Luthje S, Bottger M . (2006). Occurrence of laccases in lichenized ascomycetes of the Peltigerineae. Mycol Res 110: 846–853.
Lawrey JD . (1995). Lichen allelopathy – a review. Allelopathy ACS Symposium Series 582: 26–38.
Legendre P, Anderson MJ . (1999). Distance-based redundancy analysis: testing multispecies responses in multifactorial ecological experiments. Ecol Monogr 69: 1–24.
Leininger S, Urich T, Schloter M, Schwark L, Qi J, Nicol GW et al. (2006). Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 442: 806–809.
Li X, He Z, Zhou J . (2005). Selection of optimal oligonucleotides probes for microarrays using multiple criteria, global alignment and parameter estimation. Nucleic Acids Res 33: 6114–6123.
Line MA . (1992). Nitrogen-fixation in the sub-antarctic Macquarie Island. Polar Biol 11: 601–606.
Lingoes JC . (1971). Some boundary conditions for a monotone analysis of symmetric matrices. Psychometrika 36: 195–203.
Mancinelli RL . (1995). The regulation of methane oxidation in soil. Annu Rev Microbiol 49: 581–605.
Mancinelli RL, Shulls WA, Mckay CP . (1981). Methanol-oxidizing bacteria used as an index of soil methane content. Appl Environ Microb 42: 70–73.
Martinez-Romero E . (2001). Dinitrogen-fixing prokaryotes. In: Dworkin, M (ed). The Prokaryotes: an Evolving Electronic Resource for the Microbiological Community. Springer-Verlag: New York.
Mataloni G, Tell G, Wynn-Williams DD . (2000). Structure and diversity of soil algal communities from Cierva Point (Antarctic Peninsula). Polar Biol 23: 205–211.
Mayer AM, Staples RC . (2002). Laccase: new functions for an old enzyme. Phytochemistry 60: 551–565.
Melick DR, Bolter M, Moller R . (1994). Rates of soluble carbohydrate utilization in soils from the Windmill Islands Oasis, Wilkes Land, continental Antarctica. Polar Biol 14: 59–64.
Melick DR, Seppelt RD . (1992). Loss of soluble carbohydrates and changes in freezing-point of Antarctic bryophytes after leaching and repeated freeze-thaw cycles. Antarct Sci 4: 399–404.
Pandey KD, Kashyap AK, Gupta RK . (1992). Nitrogen-fixation by Cyanobacteria associated with moss communities in Schirmacher-Oasis, Antarctica. Isr J Bot 41: 187–198.
Philippot L, Piutti S, Martin-Laurent F, Hallet S, Germon JC . (2002). Molecular analysis of the nitrate-reducing community from unplanted and maize-planted soils. Appl Environ Microbiol 68: 6121–6128.
Pugh GJF, Allsopp D . (1982). Microfungi on Signy Island, south Orkney Islands. Br Antarct Surv Bull 57: 55–67.
Quesada A, Vincent WF, Lean DRS . (1999). Community and pigment structure of Arctic cyanobacterial assemblages: the occurrence and distribution of UV-absorbing compounds. FEMS Microbiol Ecol 28: 315–323.
Reddy CA . (1984). Physiology and biochemistry of lignin degradation. In: Klug MJ, Reddy CA (eds). Current Perspectives in Microbial Ecology. Proceedings of the Third International Symposium on Microbial Ecology. American Society for Microbiology: Washington, DC, pp 558–571.
Rhee SK, Liu XD, Wu LY, Chong SC, Wan XF, Zhou JZ . (2004). Detection of genes involved in biodegradation and biotransformation in microbial communities by using 50-mer oligonucleotide microarrays. Appl Environ Microbiol 70: 4303–4317.
Rosch C, Bothe H . (2005). Improved assessment of denitrifying, N2-fixing, and total-community bacteria by terminal restriction fragment length polymorphism analysis using multiple restriction enzymes. Appl Environ Microbiol 71: 2026–2035.
Roser DJ, Melick DR, Ling HU, Seppelt RD . (1992). Polyol and sugar content of terrestrial plants from continental Antarctica. Antarct Sci 4: 413–420.
Rozema J, Noordijk AJ, Broekman RA, van Beem A, Meijkamp BM, de Bakker NVJ et al. (2001). Poly)phenolic compounds in pollen and spores of Antarctic plants as indicators of solar UV-B – A new proxy for the reconstruction of past solar UV-B? Plant Ecol 154: 11–26.
Schlensog M, Pannewitz S, Green TGA, Schroeter B . (2004). Metabolic recovery of continental antarctic cryptogams after winter. Polar Biol 27: 399–408.
Schnell S, King GM . (1994). Mechanistic analysis of ammonium inhibition of atmospheric methane consumption in forest soils. Appl Environ Microb 60: 3514–3521.
Shapleigh JP . (2000). The denitrifying prokaryotes. In: Dworkin M (ed). The Prokaryotes: an Evolving Electronic Resource for the Microbiological Community. Springer-Verlag: New York.
Shaw MR, Harte J . (2001). Response of nitrogen cycling to simulated climate change: differential responses along a subalpine ecotone. Glob Change Biol 7: 193–210.
Smith RIL . (1994). Species-diversity and resource relationships of South Georgian fungi. Antarct Sci 6: 45–52.
Smith VR, Steenkamp M . (1992). Soil macrofauna and nitrogen on a sub-Antarctic Island. Oecologia 92: 201–206.
Solheim B, Wiggen H, Roberg S, Spaink HP . (2004). Associations between Arctic cyanobacteria and mosses. Symbiosis 37: 169–187.
Strous M, Fuerst JA, Kramer EHM, Logemann S, Muyzer G, van de Pas-Schoonen KT et al. (1999). Missing lithotroph identified as new Planctomycete. Nature 400: 446–449.
Tearle PV . (1987). Cryptogamic carbohydrate release and microbial response during freeze-thaw cycles in Antarctic fellfield fines. Soil Biol Biochem 19: 381–390.
ter Braak CJF, Šmilauer P . (2002). CANOCO Reference Manual and CanoDraw for Windows User's Guide: Software for Canonical Community Ordination (version 4.5). Microcomputer Power: Ithaca, NY.
Throbäck IN, Enwall K, Jarvis A, Hallin S . (2004). Reassessing PCR primers trageting nirS, nirK and nosZ genes for community surveys of denitrifying bacteria with DGGE. FEMS Microbiol Ecol 49: 401–417.
Thurston CF . (1994). The structure and function of fungal laccases. Microbiology 140: 19–26.
Tiquia SM, Wu LY, Chong SC, Passovets S, Xu D, Xu Y et al. (2004). Evaluation of 50-mer oligonucleotide arrays for detecting microbial populations in environmental samples. Biotechniques 36: 664–675.
van der Wal A, van Veen JA, Pijl AS, Summerbell RC, de Boer W . (2006). Constraints on development of fungal biomass and decomposition processes during restoration of arable sandy soils. Soil Biol Biochem 38: 2890–2902.
Verdnick D, Handran S, Pickett S . (2002). Key considerations for accurate microarray scanning and image analysis. In: Kamberova G (ed). DNA Image Analysis: Nuts and Bolts. DNA Press: Salem, MA, pp 83–98.
Vincent WF . (1988). Microbial Ecosystems of Antarctica. Cambridge University Press: Cambridge.
Vishniac HS . (1993). The microbiology of Antarctic soils. In: Friedmann EL (ed). Antarctic Microbiology. Wiley-Liss Inc.: New York, NY, pp 297–341.
Walton DWH . (1985). Cellulose decomposition and its relationship to nutrient cycling at South Georgia. In: Siegfried WR, Condy PR and Laws RM (eds). Antarctic Nutrient Cycling and Food Webs. Proceedings of the 4th SCAR Symposium on Antarctic Biology. Springer-Verlag: Berlin, pp 192–199.
Williamson N, Brian P, Wellington EMH . (2000). Molecular detection of bacterial and streptomycete chitinases in the environment. Antonie Van Leeuwenhoek 78: 315–321.
Wilson KH, Wilson WJ, Radosevich JL, DeSantis TZ, Viswanathan VS, Kuczmarski TA et al. (2002). High-density microarray of small-subunit ribosomal DNA probes. Appl Environ Microbiol 68: 2535–2541.
Wu LY, Liu X, Schadt CW, Zhou JZ . (2006). Microarray-based analysis of subnanogram quantities of microbial community DNAs by using whole-community genome amplification. Appl Environ Microb 72: 4931–4941.
Yergeau E, Bokhorst S, Huiskes AHL, Boschker HTS, Aerts R, Kowalchuk GA . (2007b). Size and structure of bacterial, fungal and nematode communities along an Antarctic environmental gradient. FEMS Microbiol Ecol 59: 436–451.
Yergeau E, Newsham KK, Pearce DA, Kowalchuk GA . (2007a). Patterns of bacterial diversity across a range of Antarctic terrestrial habitats. Environ Microbiol (in review).
Zhou J . (2003). Microarrays for bacterial detection and microbial community analysis. Curr Opin Microbiol 6: 288–294.
Zumft WG . (1997). Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev 61: 533–616.
Acknowledgements
This work was supported by Dutch Science Organization (NWO) grant 851.20.018 (awarded to Rien Aerts and GAK) and by The United States Department of Energy under the Genomics: GTL program through the Virtual Institute of Microbial Stress and Survival (VIMSS; http://vimss.lbl.gov), and Environmental Remediation Science Program (JZ). EY was partly supported by a Fonds Québécois pour la Recherche sur la Nature et les Technologies (FQRNT) scholarship. We are grateful to Stef Bokhorst, Merlijn Janssens and Kat Snell for sample collection and to Wietse de Boer and Wiecher Smant for soil analyses. The British Antarctic Survey, especially Pete Convey, is thanked for supporting field work. This is NIOO-KNAW publication 4031.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website (http://www.nature.com/ismej)
Supplementary information
Rights and permissions
About this article
Cite this article
Yergeau, E., Kang, S., He, Z. et al. Functional microarray analysis of nitrogen and carbon cycling genes across an Antarctic latitudinal transect. ISME J 1, 163–179 (2007). https://doi.org/10.1038/ismej.2007.24
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2007.24
Keywords
This article is cited by
-
Diversity, community structure, and abundance of nirS-type denitrifying bacteria on suspended particulate matter in coastal high-altitude aquaculture pond water
Scientific Reports (2024)
-
Arthropods, nematodes, fungi, and bacteria associated with penguin carrion in Barton Peninsula, King George Island, Antarctica
Polar Biology (2024)
-
Morphological and physiological adaptations of psychrophilic Pseudarthrobacter psychrotolerans YJ56 under temperature stress
Scientific Reports (2023)
-
Rapid growth rate responses of terrestrial bacteria to field warming on the Antarctic Peninsula
The ISME Journal (2023)
-
Microbial Catabolic Activity: Methods, Pertinence, and Potential Interest for Improving Microbial Inoculant Efficiency
Microbial Ecology (2023)
