
Abstract
Fibroblast growth factor 23 (FGF23) is a hormone that is mainly secreted by osteocytes and osteoblasts in bone. The critical role of FGF23 in mineral ion homeostasis was first identified in human genetic and acquired rachitic diseases and has been further characterised in animal models. Recent studies have revealed that the levels of FGF23 increase significantly at the very early stages of chronic kidney disease (CKD) and may play a critical role in mineral ion disorders and bone metabolism in these patients. Our recent publications have also shown that FGF23 and its cofactor, Klotho, may play an independent role in directly regulating bone mineralisation instead of producing a systematic effect. In this review, we will discuss the new role of FGF23 in bone mineralisation and the pathophysiology of CKD-related bone disorders.
Similar content being viewed by others
Introduction
Fibroblast growth factor (FGF) 23, a member of the FGF19 subfamily of FGFs, plays a key role in balancing mineral ion homeostasis and bone mineralisation.1,2,3,4 FGF23 was discovered due to its characterisation as a cause of autosomal dominant hypophosphatemic rickets4 and as the ectopically overproduced phosphaturic factor responsible for tumour-induced osteomalacia.5 Patients with such diseases suffer from impaired bone mineralisation and hypophosphataemia associated with a phosphate-wasting syndrome caused by impaired renal phosphate reabsorption and unexpectedly low levels of calcitriol.6 Observations in later studies allowed the identification of FGF23 as a key factor in the physiological regulation of phosphate and implied that it was involved in associated disorders of phosphate and bone metabolism.1,4,7
FGF23 is a 32-kDa protein that is mainly secreted by osteocytes and osteoblasts in bone. Human FGF23 consists of 251 amino acids and includes a signal peptide composed of 24 amino acids in the N-terminal portion of the protein. The half-life of intact FGF23 is 0.5–1 h in tumour-induced osteomalacia patients.8,9 The major target of FGF23 is the kidney, where it downregulates the luminal expression of sodium-phosphate cotransporters in the proximal tubule to stimulate phosphaturia.10 It also inhibits 25-hydroxyvitamin D-1α-hydroxylase and stimulates 24-hydroxylase to suppress the production of 1,25-dihydroxyvitamin D (1,25-(OH)2D).11
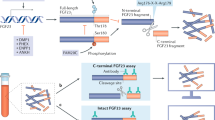
Circulating levels of FGF23 can be measured by an intact FGF23 assay, which captures intact FGF23 and C-terminal fragments. Because inactive C-terminal fragments may accumulate in patients with end-stage renal disease, we do not know whether the C-terminal FGF23 assay can provide comparable sensitivity to the intact FGF23 assay in such cases.12
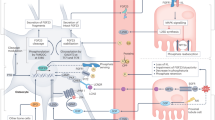
FGF23–FGF receptor–Klotho complex
To date, five distinct membrane FGF receptors (FGFRs), including FGFR1, FGFR2, FGFR3, FGFR4, and FGFR5, which belong to the tyrosine kinase superfamily, have been identified in vertebrates.13,14,15,16
Klotho, a single-pass transmembrane protein, is the cofactor implicated in the binding and activation of FGFRs by FGF23. This discovery occurred during a serendipitous experiment that examined a mouse strain that displays a premature-ageing disorder.17 Verifying that the phenotypes of Klotho-deficient mice were replicated in FGF23-deficient mice was the first step in identifying the presumptive cofactor required for FGFR binding by FGF23.18
The action of FGF23 is mediated by binding to FGF cell-surface receptors, including several types of FGFRs such as FGFR1, 3c and 4, and the FGF23 coreceptor, α-Klotho.19 FGF23 binds more robustly to FGFRs in the presence of Klotho and triggers intracellular signalling pathways that mediate its biological function. This finding can explain the restricted, tissue-specific action of FGF23, which may be secondary to the relatively limited expression of α-Klotho in specific tissues, including the parathyroid, kidney and pituitary gland. Circulating α-Klotho transduces FGF23 activity in an in vitro system at a lower level than the membrane-bound form.20 FGFs commonly signal through the extracellular signal-regulated kinases/early growth response protein 1 pathway.19
Production of FGF23
FGF23 is mainly secreted by osteocytes,21,22,23 which are cells embedded in the mineralised bone matrix that are connected to the cells outside the bone through a bone fluid-filled lacunocanalicular system.24 FGF23 shows the highest expression levels in bone, especially in osteocytes.25,26 The expression of FGF23 is increased in the osteocytes of patients with hypophosphataemic rickets and chronic kidney disease (CKD) relative to control subjects.27,28
Young and mature osteocytes regulate mineralisation and phosphate homeostasis mainly through the release of FGF23. They also regulate biomineralisation and FGF23 signalling via molecules that include phosphate-regulating genes with homologies to endopeptidases on the X chromosome (PHEX), dentin matrix protein-1 (DMP1) and matrix extracellular phosphor-glycoprotein (MEPE). PHEX, DMP1 and MEPE are all highly expressed in osteocytes.29,30,31 Mutations in PHEX can cause X-linked hypophosphataemic rickets, while mutations in DMP1 can cause autosomal recessive hypophosphataemic rickets.25,29 If the function of DMP1 or PHEX becomes altered, FGF23 can become elevated in osteocytes and the circulation, leading to the excretion of phosphate by the kidney, resulting in osteomalacia and rickets. Loss-of-function mutations and knock-out murine models have been constructed for PHEX, DMP1 and MEPE.25,29,32,33,34 In recent studies, Dmp1-null and Hyp mouse models show enhanced FGFR signalling compared with wild-type (WT) control mice, and the inhibition of FGFR signalling results in a reduction of FGF23 in the bone marrow stromal cells from both mouse models.35 These data suggest that the inhibitory effects of DMP1 and PHEX are mediated by FGFR signalling in the osteocytes and that FGFRs play a role in endocrine signalling through the osteocytes.
1,25(OH)2D can induce the expression of FGF23 in osteocytes,36,37 and parathyroid hormone (PTH) may also regulate FGF23 levels. FGF23 mRNA expression in the calvaria and serum FGF23 levels are increased in mice after the infusion of PTH.38
The extent of glycosylation and proteolytic processing can also regulate FGF23 activity at the protein level. Intact FGF23 is protected against furin-mediated cleavage to generate the C-terminal form of the peptide by glycosylation.
In different disease states, the location of the osteocytes that release FGF23 may vary. FGF23 is mainly released by the osteocytes of both cortical and trabecular bone in mouse models of hereditary hypophosphataemia.26,36 However, in a study conducted on the Col4a3-null mouse, researchers found different results. FGF23 was primarily expressed in trabecular osteocytes in this study, and significant increases in FGF23 gene expression in the bone did not occur until the later stages of CKD. The post-transcriptional processing, excretion, or breakdown of FGF23 may be altered during the early stages of CKD because FGF23 becomes elevated during this period, and osteocytes are the main site of FGF23 production.39
FGF23-associated bone diseases
FGF23-associated bone diseases refer to a group of conditions in which the function and/or amount of FGF23 is abnormal.40 This abnormality may consist of primary or secondary changes in FGF23 function (Table 1).
In cases of high FGF23 levels, patients with autosomal dominant hypophosphatemic rickets,41 X-linked hypophosphataemia,29 oncogenic osteomalacia,42 autosomal recessive hypophosphataemia, autosomal recessive hypophosphataemic rickets and fibrous dysplasia23 have the common characteristics of elevated FGF23 in the serum and hypophosphataemia. Most changes in FGF23 are non-PTH/vitamin D-dependent. Congenital or acquired dysfunction of the reabsorption of phosphorus in the kidney tubules and vitamin D deficiency syndrome are the most common causes of non-genetic hypophosphataemia. All types of hypophosphatemic rickets share the characteristic of excess or overactive FGF23,43,44 and such changes in FGF23 are most likely caused by a mutation in PHEX or MEPE.45,46,47,48 Because PTH stimulates the secretion of FGF23, high FGF23 syndrome, chronic nephropathy and cardiovascular diseases are usually aggravated by secondary parathyroidism.
Low FGF23 conditions can be divided into primary and secondary conditions. Primary low FGF23 syndrome and FGF23 deficiency are commonly found in tumoral calcinosis49 and hyperostosis hyperphosphataemia syndrome. Mutations of FGF23, Klotho, GALNT3 or SAMD9 cause an inactivating mutation of FGF23. Secondary low FGF23 syndrome is mainly accompanied by normal or decreased blood phosphorus and elevated 1,25(OH)2D and found in patients with a low phosphorus diet, mutated vitamin D receptors, mutated 1α-hydroxylase, mutated or deficient NaPi-2a or mutated NaPi-2c.
FGF23 inhibits bone mineralisation
FGF23 itself is an inhibitor of mineralisation, but whether it acts directly or indirectly is not yet known.50 Wang et al.50 showed that adenoviral overexpression of FGF23 in rat calvarial cells in vitro inhibits bone mineralisation independent of its systemic effects on phosphate homeostasis. Our group and others have also demonstrated that FGF23 treatment of primary calvarial osteoblast cultures from WT mice or from the osteoblastic MC3T3-E1 cell line leads to an inhibition of mineralisation,51,52 thus showing an effect on mineralisation independent of circulating factors.
Previously, our group and others have shown that mice lacking FGF23 function (Fgf23−/−) exhibit a phenotype of biochemical disorders including hyperphosphataemia, hypercalcaemia, high serum 1,25(OH)2D levels and decreased serum PTH levels.49,53,54 Despite the presence of a high serum mineral ion content,55,56 Fgf23−/− mice present with severe defects in skeletal mineralisation (osteomalacia/osteoidosis). The reason that this reduced skeletal mineralisation occurs in the presence of high serum calcium and phosphate is largely unknown.
Recently, we discovered that the expression of osteopontin (OPN), a well-known inhibitor of bone mineralisation,57 is substantially elevated in bone, suggesting a possible explanation for the defective mineralisation in the bones of Fgf23−/− mice.58 Both in situ hybridisation and immunohistochemistry were performed on femur sections of 6-week-old animals and showed enhanced OPN signal in the bones of Fgf23−/− mice. Moreover, to examine the accumulation of OPN at a higher resolution, we performed transmission electron microscopy after immunogold labelling for OPN in undecalcified calvarial bone. Compared with the moderate extent of immunolabelling in WT bone, seen as patches of gold particles dispersed throughout the matrix and surrounding osteocyte lacunae, the bones of Fgf23−/− mice were intensely labelled at several locations. Abundant gold particle labelling was observed over the aborted mineralisation foci in the osteoids, at the sharply demarcated mineralisation front, and immediately lining the lacunar wall surrounding the osteocytes. Osteocytes found in these heavily OPN-labelled regions of osteoid bone matrix showed prominent secretory granules that were intensely labelled for OPN, indicating the local secretion of this protein by bone cells. Furthermore, we demonstrated that ablation of Opn (Spp1) from Fgf23−/− mice significantly ameliorated the osteomalacia. Collectively, these findings indicate that increased OPN levels are responsible, in part, for the skeletal mineralisation defect observed in Fgf23−/− mice.
As mentioned above, FGF23 requires Klotho for its actions. In the absence of Klotho, FGF23 has a very low affinity for FGFR1 and cannot induce signal transduction (phosphorylation).59,60,61 The direct effect of FGF23 on bone mineralisation was unclear before Klotho was detected in the osteoblastic cell linage.62 This expression pattern was confirmed by our recent study, which showed that Klotho is expressed in both cultured osteoblasts and isolated cortical bone, although the level is much lower than that of the kidney.63
In addition to being a cofactor of FGF23, Klotho may play a specific function in osteoblasts. Kl−/− and Fgf23−/− mice share very similar phenotypes.64 Klotho knockout (Kl−/−) mice have elevated bone volume compared with the normal bone volume of Fgf23−/− mice.63,65,66 Cultured osteoblasts isolated from Kl−/− pups at the age of 2 days showed markedly impaired mineralisation, suggesting that Klotho may play a specific role in osteoblastic mineralisation. Interestingly, we recently showed that deletion of PTH might be responsible for the normalisation of the increased Opn levels in Kl−/− mice and subsequently rescue the mineralisation defect, but this same deletion did not improve the defect in Fgf23−/− animals.56 An osteoblast-specific Klotho knockout mouse model may be needed to determine the independent role of Klotho in skeletal mineralisation.
FGF 23 and CKD bone diseases
FGF23 levels increase progressively during the decline of renal function.67,68 Several studies of patients with CKD have demonstrated that the levels of circulating FGF23 begin to increase early in the course of kidney dysfunction.67,68,69,70,71 The expression levels of FGF23 in osteocytes increase,28 which provided an excellent explanation for the observations that the levels of 1,25(OH)2D begin to decline and the levels of PTH progressively increase early in the course of CKD.72 The precise mechanism by which FGF23 secretion is stimulated in response to the phosphate load is not yet clear. Studies of healthy individuals have shown that several days of a high-phosphate or low-phosphate diet results in an increase or decrease in FGF23 levels, respectively;73,74 however, FGF23 does not respond rapidly to dietary phosphate loading or intravenous administration of phosphate.75,76 Despite a significant reduction in serum phosphate levels, a session of dialysis did not decrease serum FGF23 levels in patients receiving haemodialysis,77 and elevated FGF23 levels persist for weeks to months after kidney transplantation.78,79 Together, these data suggest that FGF23 secretion is regulated by chronic phosphate load.
Normophosphataemia is maintained by FGF23 and PTH before patients reach end stage renal disease (ESRD). However, renal Klotho expression is decreased during the progression of CKD,80 leading to a reduction in the kidney's ability to excrete urinary phosphate, which will finally overcome the compensatory effects of these phosphaturia hormones. Such processes lead to an increase in serum phosphate levels, a progressive reduction in 1,25(OH)2D levels and stimulated PTH secretion. Therefore, patients with ESRD commonly suffer from hyperphosphataemia, decreased levels of 1,25(OH)2D and secondary hyperparathyroidism.72 FGF23 levels will increase markedly and often reach 100- to 1 000-fold above the normal range by the time patients start dialysis.81,82,83,84,85,86,87,88,89,90,91,92,93,94,95 Hyperphosphataemia, chronic phosphate retention, PTH levels and vitamin D will increase serum FGF23 levels, resulting in extremely high FGF23 levels.38,92,94,96,97
Elevated FGF23 levels have been linked by numerous reports to the main adverse clinical outcomes in CKD, such as progression to ESRD, cardiovascular disease and death. In a 1∶1 case–control study of 400 participants in a prospective cohort of patients with incident ESRD, elevated levels of FGF23 were independently associated with a greater risk of mortality.81 This observations helped support the concept that elevated FGF23 is not just a biomarker of phosphate-mediated cardiovascular toxicity, but is directly toxic itself.98 Another study of 219 prevalent haemodialysis patients confirmed that elevated FGF23 levels were independently associated with a greater risk of mortality during ESRD.82 Elevated FGF23 levels were independently associated with greater subsequent risks of mortality, allograft loss and composite reactions in a study of 984 prevalent kidney transplant recipients with a median transplant age at enrolment of 6 years.99
The mechanisms underlying the association of FGF23 with poor outcomes have not been established, but several possible explanations for these mechanisms have been proposed. One possible explanation is the link between inflammation and FGF23. Inflammation is common in CKD/ESRD and is associated with significantly poorer outcomes.100,101 FGF23 increases the production of inflammatory markers such as lipocalin-2, transforming growth factor-beta and tumour necrosis factor-alpha.102 In the future, the clinical relevance of the effects of FGF23 on inflammation should be tested. In a series of experiments from a single group of investigators, FGF23 induced left ventricular hypertrophy (LVH) in vitro and in experimental animals by inducing the molecular mechanisms that are typical of pathological LVH.103 These experiments challenge the prevailing paradigm that the effects of FGF23 on FGFR are weak without Klotho, even at high concentrations of FGF23, and demonstrate that FGF23 may exert direct effects on organs that do not express α-Klotho; however, more testing is needed to confirm such unorthodox hypotheses. Another mechanism could be that FGF23 acts to suppress vitamin D metabolism. By lowering vitamin D levels, FGF23 could be instrumental in producing adverse consequences, which would, under this paradigm, be related to the multiple and complex end-organ effects of low vitamin D, such as the activation of the RAAS, higher blood pressure, vascular calcification, inflammation, infections and malignancies.104 Additionally, low vitamin D levels have shown a strong association with general adverse outcomes in patients with CKD and ESRD.105,106,107
The association between FGF23 and bone in the case of CKD differs greatly from that in general cases. High circulating levels of FGF23 in paediatric dialysis patients are associated with improved indices of skeletal mineralisation in a cross-sectional analysis of 49 paediatric dialysis patients with secondary hyperparathyroidism.108 A study of FGF23, DMP1 and MEPE expression in the bone tissue of 32 paediatric and young adult patients with CKD was performed to confirm this association and demonstrated that both FGF23 and DMP1 expression were upregulated in trabecular bone in early CKD, while MEPE expression remained unchanged from normal control levels. During all stages of CKD, the expression level of bone FGF23 was directly correlated with bone DMP1 expression, and the expression of each was inversely related to osteoid accumulation. However, MEPE expression was not related to skeletal mineralisation, but was inversely related to bone volume. The simultaneous increase in both DMP1 and FGF23 expression appears to contradict previous data suggesting that DMP1 suppresses FGF23 expression, but other data have suggested that the over-expression of DMP1 does not suppress FGF23 expression.109 Additionally, DMP1 promoter activity increases in response to increasing phosphate concentrations.110 Therefore, the simultaneous increase in bone DMP1 and FGF23 expression may reflect the increasing phosphate burden associated with progressive renal failure.
To date, the pathological role of elevated FGF23 in CKD remains unclear. Researchers have developed a monoclonal FGF23 antibody to evaluate the impact of chronic FGF23 neutralisation on chronic kidney disease-mineral and bone disorder (CKD–MBD).111 CKD–MBD rats fed a high-phosphate diet were treated with low or high doses of FGF23-Ab or an isotype control antibody. Neutralisation of FGF23 led to sustained reductions in secondary HPT, including decreased parathyroid hormone; increased vitamin D; increased serum calcium; and normalisation of bone markers such as cancellous bone volume, trabecular number, osteoblast surface area, osteoid surface area and bone formation rate. However, dose-dependent increases in serum phosphate and aortic calcification associated with an increased risk of mortality in CKD–MBD rats treated with FGF23-Ab were observed. Thus, the mineral disturbances caused by neutralisation of FGF23 limit the efficacy of the FGF23-Ab and likely contribute to the increased mortality observed in this CKD–MBD rat model.111 Further studies are needed to determine whether neutralising FGF23 in CKD could ameliorate the proposed deleterious extrarenal effects of supraphysiological FGF23 on the cardiovascular system while obviating the impairments of mineral homeostasis.
References
Shimada T, Kakitani M, Yamazaki Y et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest 2004; 113 (4): 561–568.
Sitara D, Razzaque MS, Hesse M et al. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol 2004; 23 (7): 421–432.
Bergwitz C, Jüppner H . Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med 2010; 61: 91–104.
ADHR Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 2000; 26 (3): 345–348.
Shimada T, Mizutani S, Muto T et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A 2001; 98 (11): 6500–6505.
Fukumoto S, Yamashita T . FGF23 is a hormone-regulating phosphate metabolism–unique biological characteristics of FGF23. Bone 2007; 40 (5): 1190–1195.
Garringer HJ, Fisher C, Larsson TE et al. The role of mutant UDP-N-acetyl-alpha-D-galactosamine-polypeptide N-acetylgalactosaminyltransferase 3 in regulating serum intact fibroblast growth factor 23 and matrix extracellular phosphoglycoprotein in heritable tumoral calcinosis. J Clin Endocrinol Metab 2006; 91 (10): 4037–4042.
Khosravi A, Cutler CM, Kelly MH et al. Determination of the elimination half-life of fibroblast growth factor-23. J Clin Endocrinol Metab 2007; 92 (6): 2374–2377.
Takeuchi Y, Suzuki H, Ogura S et al. Venous sampling for fibroblast growth factor-23 confirms preoperative diagnosis of tumor-induced osteomalacia. J Clin Endocrinol Metab 2004; 89 (8): 3979–3982.
Shimada T, Urakawa I, Yamazaki Y et al. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun 2004; 314 (2): 409–414.
Shimada T, Hasegawa H, Yamazaki Y et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 2004; 19 (3): 429–435.
Weber TJ, Liu S, Indridason OS et al. Serum FGF23 levels in normal and disordered phosphorus homeostasis. J Bone Miner Res 2003; 18 (7): 1227–1234.
Belov AA, Mohammadi M . Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology. Cold Spring Harb Perspect Biol 2013; 5 (6): pii:a015958.
Coutts JC, Gallagher JT . Receptors for fibroblast growth factors. Immunol Cell Biol 1995; 73 (6): 584–589.
Ornitz DM, Xu J, Colvin JS et al. Receptor specificity of the fibroblast growth factor family. J Biol Chem 1996; 271 (25): 15292–15297.
Sleeman M, Fraser J, McDonald M et al. Identification of a new fibroblast growth factor receptor, FGFR5. Gene 2001; 271 (2): 171–182.
Kuro-o M, Matsumura Y, Aizawa H et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997; 390 (6655): 45–51.
Razzaque MS, Lanske B . Hypervitaminosis D and premature aging: lessons learned from Fgf23 and Klotho mutant mice. Trends Mol Med 2006; 12 (7): 298–305.
Urakawa I, Yamazaki Y, Shimada T et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006; 444 (7120): 770–774.
Kurosu H, Ogawa Y, Miyoshi M et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 2006; 281 (10): 6120–6123.
Yoshiko Y, Wang H, Minamizaki T et al. Mineralized tissue cells are a principal source of FGF23. Bone 2007; 40 (6): 1565–1573.
Mirams M, Robinson BG, Mason RS et al. Bone as a source of FGF23: regulation by phosphate? Bone 2004; 35 (5): 1192–1199.
Riminucci M, Collins MT, Fedarko NS et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest 2003; 112 (5): 683–692.
Parfitt AM . The cellular basis of bone turnover and bone loss: a rebuttal of the osteocytic resorption—bone flow theory. Clin Orthop Relat Res 1977; (127): 236–247.
Feng JQ, Ward LM, Liu S et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet 2006; 38 (11): 1310–1315.
Liu S, Zhou J, Tang W et al. Pathogenic role of Fgf23 in Dmp1-null mice. Am J Physiol Endocrinol Metab 2008; 295 (2): E254–E261.
Liu S, Rowe PS, Vierthaler L et al. Phosphorylated acidic serine-aspartate-rich MEPE-associated motif peptide from matrix extracellular phosphoglycoprotein inhibits phosphate regulating gene with homologies to endopeptidases on the X-chromosome enzyme activity. J Endocrinol 2007; 192 (1): 261–267.
Pereira RC, Juppner H, Azucena-Serrano CE et al. Patterns of FGF-23, DMP1, and MEPE expression in patients with chronic kidney disease. Bone 2009; 45 (6): 1161–1168.
A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nat Genet 1995; 11 (2): 130–136.
Thompson DL, Sabbagh Y, Tenenhouse HS et al. Ontogeny of Phex/PHEX protein expression in mouse embryo and subcellular localization in osteoblasts. J Bone Miner Res 2002; 17 (2): 311–320.
Nampei A, Hashimoto J, Hayashida K et al. Matrix extracellular phosphoglycoprotein (MEPE) is highly expressed in osteocytes in human bone. J Bone Miner Metab 2004; 22 (3): 176–184.
Rowe PS, Oudet CL, Francis F et al. Distribution of mutations in the PEX gene in families with X-linked hypophosphataemic rickets (HYP). Hum Mol Genet 1997; 6 (4): 539–549.
Strom TM, Francis F, Lorenz B et al. Pex gene deletions in Gy and Hyp mice provide mouse models for X-linked hypophosphatemia. Hum Mol Genet 1997; 6 (2): 165–171.
Gowen LC, Petersen DN, Mansolf AL et al. Targeted disruption of the osteoblast/osteocyte factor 45 gene (OF45) results in increased bone formation and bone mass. J Biol Chem 2003; 278 (3): 1998–2007.
Martin A, Liu S, David V et al. Bone proteins PHEX and DMP1 regulate fibroblastic growth factor Fgf23 expression in osteocytes through a common pathway involving FGF receptor (FGFR) signaling. FASEB J 2011; 25 (8): 2551–2562.
Liu S, Zhou J, Tang W et al. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab 2006; 291 (1): E38–E49.
Woo SM, Rosser J, Dusevich V et al. Cell line IDG-SW3 replicates osteoblast-to-late-osteocyte differentiation in vitro and accelerates bone formation in vivo. J Bone Miner Res 2011; 26 (11): 2634–2646.
Lavi-Moshayoff V, Wasserman G, Meir T et al. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol Renal Physiol 2010; 299 (4): F882–F889.
Stubbs JR, He N, Idiculla A et al. Longitudinal evaluation of FGF23 changes and mineral metabolism abnormalities in a mouse model of chronic kidney disease. J Bone Miner Res 2012; 27 (1): 38–46.
Huang XL, Jiang Y, Xia WB . FGF23 and Phosphate Wasting Disorders. Bone Res 2013; 1 (2): 120–132.
ADHR Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 2000; 26 (3): 345–348.
Nelson AE, Hogan JJ, Holm IA et al. Phosphate wasting in oncogenic osteomalacia: PHEX is normal and the tumor-derived factor has unique properties. Bone 2001; 28 (4): 430–439.
Quarles LD . Role of FGF23 in vitamin D and phosphate metabolism: implications in chronic kidney disease. Exp Cell Res 2012; 318 (9): 1040–1048.
Carpenter TO . The expanding family of hypophosphatemic syndromes. J Bone Miner Metab 2012; 30 (1): 1–9.
Chong WH, Molinolo AA, Chen CC et al. Tumor-induced osteomalacia. Endocr Relat Cancer 2011; 18 (3): R53–R77.
Owen C, Chen F, Flenniken AM et al. A novel Phex mutation in a new mouse model of hypophosphatemic rickets. J Cell Biochem 2012; 113 (7): 2432–2441.
Khaliq W, Cheripalli P, Tangella K . Tumor-induced osteomalacia (TIO): atypical presentation. South Med J 2011; 104 (5): 348–350.
van der Rest C, Cavalier E, Kaux JF et al. Tumor-induced osteomalacia: the tumor may stay hidden! Clin Biochem 2011; 44 (14/15): 1264–1266.
Larsson T, Yu X, Davis SI et al. A novel recessive mutation in fibroblast growth factor-23 causes familial tumoral calcinosis. J Clin Endocrinol Metab 2005; 90 (4): 2424–2427.
Wang H, Yoshiko Y, Yamamoto R et al. Overexpression of fibroblast growth factor 23 suppresses osteoblast differentiation and matrix mineralization in vitro. J Bone Miner Res 2008; 23 (6): 939–948.
Sitara D, Kim S, Razzaque MS et al. Genetic evidence of serum phosphate-independent functions of FGF-23 on bone. PLoS Genet 2008; 4 (8): e1000154.
Shalhoub V, Ward SC, Sun B et al. Fibroblast growth factor 23 (FGF23) and alpha-klotho stimulate osteoblastic MC3T3.E1 cell proliferation and inhibit mineralization. Calcif Tissue Int 2011; 89 (2): 140–150.
Sitara D, Razzaque MS, St-Arnaud R et al. Genetic ablation of vitamin D activation pathway reverses biochemical and skeletal anomalies in Fgf-23-null animals. Am J Pathol 2006; 169 (6): 2161–2170.
Razzaque MS, Sitara D, Taguchi T et al. Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin D-mediated process. FASEB J 2006; 20 (6): 720–722.
Yuan Q, Sato T, Densmore M et al. FGF-23/Klotho signaling is not essential for the phosphaturic and anabolic functions of PTH. J Bone Miner Res 2011; 26 (9): 2026–2035.
Yuan Q, Sitara D, Sato T et al. PTH ablation ameliorates the anomalies of Fgf23-deficient mice by suppressing the elevated vitamin D and calcium levels. Endocrinology 2011; 152 (11): 4053–4061.
Sodek J, Ganss B, McKee MD . Osteopontin. Crit Rev Oral Biol Med 2000; 11 (3): 279–303.
Yuan Q, Jiang Y, Zhao X et al. Increased osteopontin contributes to inhibition of bone mineralization in FGF23-deficient mice. J Bone Miner Res 2014; 29 (3): 693–704.
Urakawa I, Yamazaki Y, Shimada T et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006; 444 (7120): 770–774.
Kurosu H, Ogawa Y, Miyoshi M et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 2006; 281 (10): 6120–6123.
Tomiyama K, Maeda R, Urakawa I et al. Relevant use of Klotho in FGF19 subfamily signaling system in vivo. Proc Natl Acad Sci U S A 2010; 107 (4): 1666–1671.
Rhee Y, Bivi N, Farrow E et al. Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone 2011; 49 (4): 636–643.
Yuan Q, Sato T, Densmore M et al. Deletion of PTH rescues skeletal abnormalities and high osteopontin levels in Klotho−/− mice. PLoS Genet 2012; 8 (5): e1002726.
Nakatani T, Sarraj B, Ohnishi M et al. In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23)-mediated regulation of systemic phosphate homeostasis. FASEB J 2009; 23 (2): 433–441.
Liu H, Fergusson MM, Castilho RM et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science 2007; 317 (5839): 803–806.
Brownstein CA, Zhang J, Stillman A et al. Increased bone volume and correction of HYP mouse hypophosphatemia in the Klotho/HYP mouse. Endocrinology 2010; 151 (2): 492–501.
Gutierrez O, Isakova T, Rhee E et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol 2005; 16 (7): 2205–2215.
Larsson T, Nisbeth U, Ljunggren O et al. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int 2003; 64 (6): 2272–2279.
Shigematsu T, Kazama JJ, Yamashita T et al. Possible involvement of circulating fibroblast growth factor 23 in the development of secondary hyperparathyroidism associated with renal insufficiency. Am J Kidney Dis 2004; 44 (2): 250–256.
Komaba H, Fukagawa M . FGF23-parathyroid interaction: implications in chronic kidney disease. Kidney Int 2010; 77 (4): 292–298.
Gutiérrez OM . Fibroblast growth factor 23, Klotho, and disordered mineral metabolism in chronic kidney disease: unraveling the intricate tapestry of events and implications for therapy. J Ren Nutr 2013; 23 (3): 250–254.
Levin A, Bakris GL, Molitch M et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int 2007; 71 (1): 31–38.
Ferrari SL, Bonjour JP, Rizzoli R . Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab 2005; 90 (3): 1519–1524.
Burnett SM, Gunawardene SC, Bringhurst FR et al. Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res 2006; 21 (8): 1187–1196.
Nishida Y, Taketani Y, Yamanaka-Okumura H et al. Acute effect of oral phosphate loading on serum fibroblast growth factor 23 levels in healthy men. Kidney Int 2006; 70 (12): 2141–2147.
Ito N, Fukumoto S, Takeuchi Y et al. Effect of acute changes of serum phosphate on fibroblast growth factor (FGF)23 levels in humans. J Bone Miner Metab 2007; 25 (6): 419–422.
Urena Torres P, Friedlander G, de Vernejoul MC et al. Bone mass does not correlate with the serum fibroblast growth factor 23 in hemodialysis patients. Kidney Int 2008; 73 (1): 102–107.
Bhan I, Shah A, Holmes J et al. Post-transplant hypophosphatemia: tertiary ‘Hyper-Phosphatoninism’? Kidney Int 2006; 70 (8): 1486–1494.
Evenepoel P, Naesens M, Claes K et al. Tertiary ‘hyperphosphatoninism’ accentuates hypophosphatemia and suppresses calcitriol levels in renal transplant recipients. Am J Transplant 2007; 7 (5): 1193–1200.
Koh N, Fujimori T, Nishiguchi S et al. Severely reduced production of klotho in human chronic renal failure kidney. Biochem Biophys Res Commun 2001; 280 (4): 1015–1020.
Gutiérrez OM, Mannstadt M, Isakova T et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 2008; 359 (6): 584–592.
Jean G, Terrat JC, Vanel T et al. High levels of serum fibroblast growth factor (FGF)-23 are associated with increased mortality in long haemodialysis patients. Nephrol Dial Transplant 2009; 24 (9): 2792–2796.
Olauson H, Qureshi AR, Miyamoto T et al. Relation between serum fibroblast growth factor-23 level and mortality in incident dialysis patients: are gender and cardiovascular disease confounding the relationship? Nephrol Dial Transplant 2010; 25 (9): 3033–3038.
Fukagawa M, Kazama JJ . With or without the kidney: the role of FGF23 in CKD. Nephrol Dial Transplant 2005; 20 (7): 1295–1298.
Kazama JJ, Sato F, Omori K et al. Pretreatment serum FGF-23 levels predict the efficacy of calcitriol therapy in dialysis patients. Kidney Int 2005; 67 (3): 1120–1125.
Nakanishi S, Kazama JJ, Nii-Kono T et al. Serum fibroblast growth factor-23 levels predict the future refractory hyperparathyroidism in dialysis patients. Kidney Int 2005; 67 (3): 1171–1178.
Nishi H, Nii-Kono T, Nakanishi S et al. Intravenous calcitriol therapy increases serum concentrations of fibroblast growth factor-23 in dialysis patients with secondary hyperparathyroidism. Nephron Clin Pract 2005; 101 (2): c94–c99.
Wesseling-Perry K, Pereira RC, Sahney S et al. Calcitriol and doxercalciferol are equivalent in controlling bone turnover, suppressing parathyroid hormone, and increasing fibroblast growth factor-23 in secondary hyperparathyroidism. Kidney Int 2011; 79 (1): 112–119.
Hansen D, Rasmussen K, Pedersen SM et al. Changes in fibroblast growth factor 23 during treatment of secondary hyperparathyroidism with alfacalcidol or paricalcitol. Nephrol Dial Transplant 2012; 27 (6): 2263–2269.
Wetmore JB, Liu S, Krebill R et al. Effects of cinacalcet and concurrent low-dose vitamin D on FGF23 levels in ESRD. Clin J Am Soc Nephrol 2010; 5 (1): 110–116.
Koizumi M, Komaba H, Nakanishi S et al. Cinacalcet treatment and serum FGF23 levels in haemodialysis patients with secondary hyperparathyroidism. Nephrol Dial Transplant 2012; 27 (2): 784–790.
Koiwa F, Kazama JJ, Tokumoto A et al. Sevelamer hydrochloride and calcium bicarbonate reduce serum fibroblast growth factor 23 levels in dialysis patients. Ther Apher Dial 2005; 9 (4): 336–339.
Sato T, Tominaga Y, Ueki T et al. Total parathyroidectomy reduces elevated circulating fibroblast growth factor 23 in advanced secondary hyperparathyroidism. Am J Kidney Dis 2004; 44 (3): 481–487.
Shigematsu T, Negi S, COLC Research Group. Combined therapy with lanthanum carbonate and calcium carbonate for hyperphosphatemia decreases serum FGF-23 level independently of calcium and PTH (COLC Study). Nephrol Dial Transplant 2012; 27 (3): 1050–1054.
Takeda Y, Komaba H, Goto S et al. Effect of intravenous saccharated ferric oxide on serum FGF23 and mineral metabolism in hemodialysis patients. Am J Nephrol 2011; 33 (5): 421–426.
López I, Rodríguez-Ortiz ME, Almadén Y et al. Direct and indirect effects of parathyroid hormone on circulating levels of fibroblast growth factor 23 in vivo. Kidney Int 2011; 80 (5): 475–482.
Liu S, Tang W, Zhou J et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol 2006; 17 (5): 1305–1315.
Jüppner H, Wolf M, Salusky IB . FGF-23: more than a regulator of renal phosphate handling? J Bone Miner Res 2010; 25 (10): 2091–2097.
Wolf M, Molnar MZ, Amaral AP et al. Elevated fibroblast growth factor 23 is a risk factor for kidney transplant loss and mortality. J Am Soc Nephrol 2011; 22 (5): 956–966.
Kalantar-Zadeh K, Kopple JD, Humphreys MH et al. Comparing outcome predictability of markers of malnutrition-inflammation complex syndrome in haemodialysis patients. Nephrol Dial Transplant 2004; 19 (6): 1507–1519.
Stenvinkel P, Lindholm B, Heimbürger M et al. Elevated serum levels of soluble adhesion molecules predict death in pre-dialysis patients: association with malnutrition, inflammation, and cardiovascular disease. Nephrol Dial Transplant 2000; 15 (10): 1624–1630.
Dai B, David V, Martin A et al. A comparative transcriptome analysis identifying FGF23 regulated genes in the kidney of a mouse CKD model. PLoS ONE 2012; 7 (9): e44161.
Faul C, Amaral AP, Oskouei B et al. FGF23 induces left ventricular hypertrophy. J Clin Invest 2011; 121 (11): 4393–4408.
Kovesdy CP, Kalantar-Zadeh K . Vitamin D receptor activation and survival in chronic kidney disease. Kidney Int 2008; 73 (12): 1355–1363.
Melamed ML, Michos ED, Post W et al. 25-hydroxyvitamin D levels and the risk of mortality in the general population. Arch Intern Med 2008; 168 (15): 1629–1637.
Mehrotra R, Kermah DA, Salusky IB et al. Chronic kidney disease, hypovitaminosis D, and mortality in the United States. Kidney Int 2009; 76 (9): 977–983.
Wolf M, Shah A, Gutierrez O et al. Vitamin D levels and early mortality among incident hemodialysis patients. Kidney Int 2007; 72 (8): 1004–1013.
Wesseling-Perry K, Pereira RC, Wang H et al. Relationship between plasma fibroblast growth factor-23 concentration and bone mineralization in children with renal failure on peritoneal dialysis. J Clin Endocrinol Metab 2009; 94 (2): 511–517.
Lu Y, Ye L, Yu S et al. Rescue of odontogenesis in Dmp1-deficient mice by targeted re-expression of DMP1 reveals roles for DMP1 in early odontogenesis and dentin apposition in vivo. Dev Biol 2007; 303 (1): 191–201.
Lu Y, Liu S, Xie Y et al. Use of the transgenic approach to determine the role of DMP1 in phosphate regulation. J Musculoskelet Neuronal Interact 2007; 7 (4): 309.
Shalhoub V, Shatzen EM, Ward SC et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J Clin Invest 2012; 122 (7): 2543–2553.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81371173), the Program for New Century Excellent Talents in University (NCET-12-0379) and Sichuan Provincial Government Grant (2013JQ0017). Publication of this manuscript is supported by Open Fund of State Key Laboratory of Oral Diseases, Sichuan University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Guo, YC., Yuan, Q. Fibroblast growth factor 23 and bone mineralisation. Int J Oral Sci 7, 8–13 (2015). https://doi.org/10.1038/ijos.2015.1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijos.2015.1
Keywords
This article is cited by
-
Linea osteoblastica-osteocitaria e produzione ormonale
L'Endocrinologo (2024)
-
Regulation of the Osteocyte Secretome with Aging and Disease
Calcified Tissue International (2023)
-
Potential influences on optimizing long-term musculoskeletal health in children and adolescents with X-linked hypophosphatemia (XLH)
Orphanet Journal of Rare Diseases (2022)
-
Fibroblast growth factor 21 (FGF21) is a sensitive marker of osteoporosis in haemodialysis patients: a cross-sectional observational study
BMC Nephrology (2021)
-
Genetic renal disease classification by hormonal axes
Pediatric Nephrology (2020)
