
Abstract
Dentin matrix protein 1 (DMP1) is essential to odontogenesis. Its mutations in human subjects lead to dental problems such as dental deformities, hypomineralization and periodontal impairment. Primarily, DMP1 is considered as an extracellular matrix protein that promotes hydroxyapatite formation and activates intracellular signaling pathway via interacting with αvβ3 integrin. Recent in vitro studies suggested that DMP1 might also act as a transcription factor. In this study, we examined whether full-length DMP1 could function as a transcription factor in the nucleus and regulate odontogenesis in vivo. We first demonstrated that a patient with the DMP1 M1V mutation, which presumably causes a loss of the secretory DMP1 but does not affect the nuclear translocation of DMP1, shows a typical rachitic tooth defect. Furthermore, we generated transgenic mice expressing NLSDMP1, in which the endoplasmic reticulum (ER) entry signal sequence of DMP1 was replaced by a nuclear localization signal (NLS) sequence, under the control of a 3.6 kb rat type I collagen promoter plus a 1.6 kb intron 1. We then crossbred the NLSDMP1 transgenic mice with Dmp1 null mice to express the NLSDMP1 in Dmp1-deficient genetic background. Although immunohistochemistry demonstrated that NLSDMP1 was localized in the nuclei of the preodontoblasts and odontoblasts, the histological, morphological and biochemical analyses showed that it failed to rescue the dental and periodontal defects as well as the delayed tooth eruption in Dmp1 null mice. These data suggest that the full-length DMP1 plays no apparent role in the nucleus during odontogenesis.
Similar content being viewed by others
Introduction
Dentin matrix protein 1 (DMP1), a member of the small integrin-binding ligand, N-linked glycoproteins family,1 plays essential roles in osteogenesis and odontogenesis. DMP1 mutations in humans result in autosomal recessive hypophosphatemic rickets-1 (ARHR1; OMIM#241520), characterized by rickets/osteomalacia, hypophosphatemia and elevated circulating fibroblast growth factor 23.2 Dmp1 null mice also develop an ARHR-like phenotype.2 The teeth of Dmp1 null mice display enlarged pulp cavities and root canals, increased thickness of the predentin zone with a reduced dentin wall, hypomineralization of dentin, abnormalities in the ultrastructure of dentinal tubules, delayed development of the third molar, breakdown of periodontal structures and other consequences.3,4,5,6
Several lines of evidence support the finding that DMP1 acts as an extracellular matrix protein. First, the initial 16 amino-acid residues of DMP1 deduced from its cDNA is a typical endoplasmic reticulum (ER) signal sequence, and this protein is found abundantly in the bone and dentinal matrix.7,8 Second, the DMP1 protein sequence contains some specific acidic clusters that control the formation of oriented calcium phosphate crystals.9,10 Gajjeraman and others11,12,13 further confirmed that both the full-length DMP1 and the COOH-terminal fragment could accelerate the nucleation of hydroxyapatite crystals. Lastly, studies from our lab and others demonstrated that DMP1 has the ability to activate the extracellular signal-regulated kinase/mitogen-activated protein kinase pathways via the αvβ3 integrin, leading to the translocation of phosphorylated cellular Jun N-terminal kinase (JNK) into the nucleus and the concomitant upregulation of transcriptional activation by phosphorylated c-Jun.3,14,15
However, in vitro studies indicated that DMP1 also might function as a transcription factor in the nucleus. George et al.16 first reported that the DMP1 protein sequence contained a functional nuclear localization signal (NLS) sequence, which was thought to be responsible for the nuclear import of full-length DMP1 in pre-odontoblast-like cell. They further demonstrated that the extracellular full-length DMP1 could be internalized via endocytosis mediated by glucose-regulated protein-78, an ER chaperone protein found on the plasma membrane of pre-odontoblasts and subsequently transported into the nucleus. Once in the nucleus, DMP1 regulates the transcription of odontoblast-specific genes such as DSPP.16,17 These investigators proposed that during the maturation of the pre-odontoblast/odontoblast, this nuclear DMP1 would be phosphorylated by casein kinase II, leading to its exportation into the extracellular matrix where it promotes hydroxyapatite formation.16 However, the role of nuclear DMP1 has been challenged by the identification of several patients who presumably have normal nuclear localization of DMP1 due to a biallelic nucleotide substitution in the DMP1 start codon (ATG to GTG, or A→G), but who display hypophosphatemic rickets.2 Therefore, whether DMP1 can enter the nucleus and function as a transcription factor in vivo needs to be further studied.
This study was aimed at determining the in vivo function, if any, of the nuclear DMP1 in odontogenesis. Our human studies revealed that an ARHR patient, with a M1V mutation at the DMP1 start codon, manifested typical rachitic dental defects. Animal studies showed that the targeted expression of NLSDMP1 in the Dmp1 null nucleus failed to rescue the Dmp1 null dental defects, suggesting that DMP1 plays no apparent role in the nuclei during odontogenesis.
Materials and methods
Human subjects
The study protocol and patient consents were reviewed and approved by the Institutional Review Boards at the University of Ottawa. Dental radiographs were obtained from the previously described individual.2
Generation of col1α1-NLSDMP1 transgenic mice
For the generation of the Col1α1-NLSDMP1 transgene, ER-entry signal sequence of the full-length mouse Dmp1 cDNA, was replaced by an NLS (PPKKKRKV) sequence (NLSDMP1). Together with a SV40 polyadenylation signal, the NLSDMP1 was cloned into the EcoR V and Sal I sites of a mammalian expression vector18 downstream of the 3.6 kb rat type I collagen promoter plus a 1.6 kb intron 1. The Col1α1-NLSDMP1 transgene was then released from the vector backbone by Sac II and Sal I and purified for pronuclear injection. Transgenic founders with a C57B/L6 background were generated at the UT Southwestern Medical Center (Dallas, TX, USA). Genotyping was conducted as previously.3,4
Targeted expression of the NLSDMP1 in Dmp1 null mice
Three of five independent transgenic lines were separately crossed with Dmp1 null (Dmp1−/−) mice to introduce the Col1α1-NLSDMP1 transgene into the Dmp1−/− mice (referred to as ‘Dmp1−/−; Col1α1-NLSDMP1’). All three transgenic lines had similar rescue effects on the Dmp1 null dental defects, so only the data obtained from line #95 are presented. The littermate Dmp1 heterozygous (Dmp1+/−) mice were used as control mice since there was no phenotypic difference between these Dmp1+/− and wild-type mice. The mice were fed with Purina rodent chow (5010; Ralston Purina, St Louis, MO, USA) containing 1% calcium, 0.67% phosphorus, and 4.4 IU of vitamin D per gram. All the animal procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Texas A&M University Baylor College of Dentistry (Dallas, TX, USA).
Histology
Animals were sacrificed at different ages (10-day-old, 3-week-old, 6-week-old and 1-year-old) and perfused with freshly prepared 4% paraformaldehyde in phosphate buffer solution (PBS) (pH 7.4). The lower jaws were dissected and decalcified, followed by standard paraffin embedding. Sections (4.5 μm) were cut for histochemistry staining and immunohistochemistry staining as previously described.2 Toluidine Blue and Sirius Red (also called ‘Direct Red 80’) were obtained from Sigma (St Louis, MO, USA). Sections were stained with Toluidine blue for 5 min, while Sirius Red for 1 h. The tartrate resistant acid phosphatase (TRAP) staining and immunohistochemistry procedures were previously described.2 Antibodies used for immunohistochemistry included anti-Dmp1-C-terminal polyclonal antibody,19 anti-rat DSP monoclonal antibody20 (also recognize DSP from mice21) and anti-Biglycan monoclonal antibody.22 After staining, sections were dehydrated, mounted, and imaged under light microscopy and/or polarized microscopy (ECLIPSE 80i; Nikon, Tokyo, Japan).
High-resolution radiography, microcomputed tomography and resin cast scanning electron microscopy
The mouse mandibles were fixed in freshly prepared 4% paraformaldehyde/PBS (pH 7.4) for two days, then dissected free of muscle and kept in 0.5% paraformaldehyde/PBS (pH 7.4). High-resolution radiographs were taken using a plain X-ray radiography system (Faxitron MX-20DC12 system; Faxitron Bioptics, Lincolnshire, IL, USA) with a digital camera (Faxitron X-Ray, Lincolnshire, IL, USA). Mesial root length and thickness of the first molar were determined based on the radiographs using ImageJ (National Institutes of Health, Bethesda, MD, USA). Microcomputed tomography (µCT) analysis was performed with the Scanco µCT35 (Scanco Medical, Bassersdorf, Switzerland) as described previously.3 For scanning electron microscopy analysis, the samples were embedded in methyl methacrylate and cut to expose the dental pulps and root canals; the surface was then polished using alumina alpha micropolish II solution (Buehler, Lake Bluff, IL, USA). For backscatter electron microscopy analysis, samples were then coated with carbon; while for the acid-etch electron microscopy analysis, samples were acid etched with 37% phosphoric acid for 5 s, 5% sodium hydrochloride for 5 min, gold-coated and then scanned by a FEI/Philips XL30 field-emission environmental scanning electron microscopy (FEI/Philips, Hillsboro, OR, USA) as described previously.23
Statistical analysis
Data analysis was performed with one-way analysis of variance (ANOVA) for multiple-group comparison. If significant differences were found with one-way ANOVA, the Bonferroni method was used to determine which groups were significantly different from others. The quantified results were presented as the mean±standard error of the mean (S.E.M.). P<0.05 was considered statistically significant.
Results
A patient with a DMP1 M1V mutation displayed deformed teeth
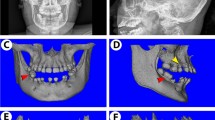
We previously reported that a DMP1 M1V mutation was identified in an ARHR patient.2 The DMP1 in this patient had a start codon mutation (A→G) resulting in the substitution of the initial methionine with valine. As a result, the original start codon lost its function and a subsequent signal acted as a new start codon for the protein translation (Figure 1a). Further analysis revealed that the M1V mutant DMP1 protein was wholly retained within the cells as the 94-kDa full-length DMP1 without cleavage, consistent with the change of translational initiation (at an internal methionine) that results in the loss of the ER-entry signal peptide.2,24 Dental radiographs (Figure 1b, upper panel) showed enlarged pulp cavities and root canals as well as thinner dentin, compared to an age-matched control (Figure 1b, lower panel). These dental defects were very similar to those of other DMP1 mutation kindreds25 and Dmp1−/−mice.4,5

Tooth phenotype in an M1V mutant ARHR patient. (a) Comparison of the amino acid sequence of the NH2-terminal region of DMP1 across species, including the M1V mutant human DMP1. (b) Dental radiographs from an adult (20+ years old) ARHR patient (upper panel) and the age-matched control (lower panel). The patient had enlarged pulp cavities and root canals and reduced dentin thickness (arrows) compared to the control. ARGR, autosomal recessive hypophosphatemic ricket; DMP1, dentin matrix protein 1; Mut, mutation; yr., year.
NLSDMP1 did not rescue the dental defects of Dmp1−/− mice
To understand whether DMP1 plays a role in the nuclei in vivo, we generated Col1α1-NLSDMP1 transgenic mice, which not only mimic the M1V mutation that leads to the suppression of its secretion, but also ensure its nuclear transportation by using an NLS sequence to replace its ER-entry signal sequence. Three of five independent lines were then crossed with the Dmp1−/− mice to introduce NLSDMP1 into the Dmp1−/− background. All three transgenic lines showed a similar phenotype, although only the results obtained from line #95 were presented. Immunohistochemistry staining of DMP1 revealed that in the control Dmp1+/− mice, endogenous DMP1 was detected in the dentinal matrix and alveolar bone matrix, as well as the cytoplasm and dendrites of odontoblasts (Figure 2a–2c), compared to no signal in the Dmp1−/− mice (Figure 2a–2c). Notably, NLSDMP1 was identified in the nuclei, cytoplasm and dendrites of the pre-odontoblasts and odontoblasts, but not in the dentinal matrix of the Dmp1−/−; Col1α1-NLSDMP1 mice (Figure 2a–2c).

Nucleus-targeted Dmp1 did not rescue the dental abnormalities of Dmp1−/− mice. (a) The Dmp1 expression was analyzed in the first molars of 3-week-old mice by immunohistochemical staining using anti-Dmp1-C-terminal antibody (signal in brown color). (b, c) Higher magnification of the boxed areas in (a) demonstrated the presence of Dmp1 in the odontolineage and dentinal matrix. (b) Note the localization of NLSDMP1 in the nuclei, cytoplasm and dendrites of the odontoblasts (arrows), but not in the dentinal matrix in the Dmp1−/−; Col1α1-NLSDMP1 mice. (c) Localization of NLSDMP1 in the nuclei of pre-odontoblasts (arrows). (d) Representative radiographs of 6-week-old mouse tooth. Note the enlarged pulp cavities and root canals (arrows) and the thinner dentin (arrowheads) in both Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 mice. (e) Toluidine Blue staining of the first molars of 3-week-old mice. Note the thinner crown dentin (arrows) in both the Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 teeth. (f) Higher magnification of the boxed area in (e). Note the widened predentin (arrows) with thinner dentin in Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 mice. (g) The toluidine blue staining of 6-week-old mouse dentin. Note the widened predentin (arrows) with reduced dentin thickness, as well as the presence of interglobular dentin (arrowheads) in Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 mice. (h) Sirius Red-stained sections imaged under a polariscope. Orange indicates larger collagen fibers, whereas green means thinner collagen fibers, such as reticular fibers. (i) Anti-DSP-antibody staining of 3-week-old mice. DSP was downregulated in the odontoblasts (arrows) and dentinal matrix (asterisks) of the Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 mice. (j) Images of the dentinal tubular system from resin-cast acid-etched electron microscopy. Note that the dendritic branches are fewer and disorganized in the Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 mice. DMP1, dentin matrix protein 1; Dn, dentin; En, enamel; P, pulp; pDn, predentin; PL, periodontal ligament; W, week.
Next, a series of experiments were performed to assess whether the targeted expression of the NLSDMP1 had rescue effects on the Dmp1 null dental defects. The representative radiographs demonstrated that, similar to the Dmp1−/− teeth, the teeth of the Dmp1−/−; Col1α1-NLSDMP1 mice teeth displayed significantly thinner dentin and shorter roots (Figure 2d, Supplementary Figure S1a and S1b, P<0.01), enlarged pulp cavities and root canals (Figure 2d). Histological analysis further confirmed that, similar to the Dmp1−/− mice, the Dmp1−/−; Col1α1-NLSDMP1 mice teeth manifested a reduced thickness of the dentin walls and an increased thickness of the pre-dentin zone (Figure 2e–2f), indicating that there was a partial failure of pre-dentin to dentin maturation. At 6 weeks of age, the expanded pre-dentin remained, and the interglobular dentin, which originated from the incompletely calcified dentinal matrices between the calcified globules, were found in both the Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 mice (Figure 2g). In addition, larger collagen fibers (Figure 2h) and high levels of biglycan (Figure 3f) were present in the dentin or pre-dentin in both the Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 mice, compared to the control mice. Furthermore, DSPP is predominantly expressed in the odontoblast and is essential for dentin mineralization,26,27 immunohistochemical staining revealed that DSP, the amino-terminal portion of DSPP, was considerably decreased in both the Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 mice (Figure 2i). Scanning electron microscopy analysis showed few dentinal tubules and branches in both Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 mice (Figure 3j). All of these data demonstrated that the NLSDMP1 transgene failed to rescue the defects in odontoblast maturation and dentinal mineralization in Dmp1−/− mice.

Periodontal structures did not improve in nucleus-targeted Dmp1. (a) Radiographs of a 1-year-old mouse tooth. Note the combined periodontal-endodontic lesions in the second molar (arrows) and the exfoliation of the second molar (arrowheads). (b, c) Occlusal and buccal photographs of 1-year-old mouse tooth. Note the periodontitis with notable loss of periodontal attachment (arrows) and the exfoliation of the second molar with alveolar bone absorption (arrowheads). (d) Sirius Red-stained image under light microscopy (3-week-old mouse molars). Dark red indicates more collagen contents; light red means less collagen contents. (e) The same sections of (d) imaged under a polariscope (3-week-old mouse molars). (f) Anti-biglycan antibody staining of 3-week-old mouse molars. The biglycan was increased in the widened predentin (arrowheads) and the matrix of alveolar bone (asterisks), but was decreased in the PDL (arrows) in Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 groups. (g, h) Backscatter electron microscopy of alveolar bone. Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 groups showed the disorganization and hypomineralization of the trabeculae. AB, alveolar bone; DMP1, dentin matrix protein 1; HET, heterozygous; KO,knockout; PDL, periodontal ligament.
NLSDMP1 did not cure the defects of periodontal structures in Dmp1−/− mice
We previously reported that Dmp1−/− mice developed an early-onset periodontal defect,3,4,5,6 including hypomineralized alveolar bone, abnormal cementum and periodontal ligament (PDL). When the Dmp1−/− mice got older, they developed typical combined periodontal-endodontic lesions or tooth exfoliation (Figure 3a), which might be due to the loss of the periodontal attachment (Figure 3b and 3c). As a result, we also analyzed the periodontal structures in different groups to identify whether NLSDMP1 rescued the periodontal defects in Dmp1−/− mice. Sirius Red staining further confirmed that the collagen contents had decreased in the PDL (Figure 3d) and that the collagen fibers appeared to be thinner, suggesting the presence of more reticular fibers (Figure 3e) in both the Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 mice, compared to the control mice. Biglycan staining also showed reduced biglycan protein in the PDL of the Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 mice (Figure 3f). Furthermore, the alveolar bone was hypomineralized and not well organized in the Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 mice (Figure 3g–3h). Collectively, these observations indicated that NLSDMP1 did not rescue the periodontal defects of the Dmp1−/− mice.
NLSDMP1 did not prevent the delay of tooth eruption in Dmp1−/− mice
Unexpectedly, we also discovered a delayed tooth eruption in the Dmp1−/− mice. At the age of 10 days, the first molars had not fully erupted and were still partially embedded in the bony crypts in both the Dmp1−/− and the Dmp1−/−; Col1α1-NLSDMP1 mice (Figure 4a and 4b). The development of the bony crypt for the third molars was also delayed in the Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 mice (Figure 4a and 4b). By the age of 3 weeks, these third molars had still not fully erupted compared to the control mice (Figure 4c and 4d). Furthermore, TRAP staining of alveolar bone showed that there was a significant decrease in osteoclast number in both Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 mice (Supplementary Figure S1c). These findings suggested that the delayed tooth eruption in the Dmp1−/− mice might be due to a decrease in osteoclast activity and thatNLSDMP1 did not rescue this defect.

Nucleus-targeted Dmp1 showed no effect on tooth eruptional delay. (a, b) Representative radiographs and μCT images of 10-day-old mouse mandibles. The bony crypts (arrows) indicate where the third molar will develop in the Dmp1+/− and Dmp1+/−; Col1α1-NLSDMP1 mice, whereas they were barely visible in the Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 mice. In addition, the occlusal surface (arrowheads) of the first molars was partially embedded in the bony crypts in both Dmp1−/− and Dmp1−/−; Col1α1-NLSDMP1 mice. (c, d) Representative radiographs and μCT images of 3-week-old mouse mandibles; note the partial eruption of the third molars (arrows). 1st, first molar; 2nd, second molar; 3rd, third molar. D, day; W, week. DMP1, dentin matrix protein 1; μCT, microcomputed tomography.
Discussion
DMP1 is actively expressed in the mandibular odontoblasts, osteoblasts and osteocytes. In vitro studies suggested that the DMP1 C-terminal contains NLS signals and can function as a transcription factor during osteogenesis/odontogenesis.16 In this study, we investigated the potential nuclear function of the full-length DMP1 in vivo in odontogenesis using a clinical case and mutant mouse models. Our results showed that (i) DMP1 mutations (M1V, resulting in a loss of the ER-entry signal peptide but with the intact DMP1 preserved in the cell) led to enlarged pulp/root canals and thin dentin; (ii) the nucleus-targeted Dmp1 transgene failed to rescue the tooth defects in Dmp1 null mice; and (ii) Dmp1 null mice unexpectedly developed delayed tooth eruption, which was not rescued by the nucleus-targeted Dmp1 transgene either. Thus, the current studies do not support the hypothesis that the full-length DMP1 has any nuclear function in vivo.
We previously reported that the re-expression of secretory full-length Dmp1 under the control of a 3.6 kb Col1α1 promoter completely rescued the skeletal defects and serum biochemical abnormalities of Dmp1−/− mice by the age of 7 weeks.23 However, when the same 3.6 kb Col1α1 promoter was used to drive the expression of nucleus-targeted NLSDMP1, different transgenic lines (Line #95 and Lin #97, Supplementary Figure S1d) consistently showed that the NLSDMP1 failed to rescue the defects of the tooth and periodontal structures or the delayed tooth eruption observed in Dmp1−/− mice. In addition, we showed that the ARHR patient with a M1V mutation, which presumably affected the secretion of DMP1 but not its nuclear localization, manifested a typical rachitic tooth defect. These mouse and human genetic studies suggest that DMP1 might have no apparent role in the nucleus in vivo.
George's group16,17 proposed that DMP1 may shuttle between the nucleus and extracellular milieu, as they observed that DMP1 contains both the functional NLS and the functional nuclear export signal sequences.16 They speculated that during osteoblast/odontoblast differentiation, the extracellular DMP1 is taken up again by the cells via glucose-regulated protein-78-mediated endocytosis and subsequently transported into the nucleus to regulate the expression of the osteoblast-/odontoblast-specific genes.17 They also proposed that during osteoblast maturation, DMP1 is phosphorylated and exported into the extracellular matrix to regulate matrix mineralization.16 In the current study, we demonstrated that NLSDMP1 is localized in the nuclei of Dmp1 null odontoblasts and that the M1V mutant DMP1 with the NLS signal in its intact molecule should be able to enter the nuclei. Therefore, both NLSDMP1 and M1V mutant DMP1 should be exported into the extracellular matrix. However, we showed that NLSDMP1 was neither found in the matrix nor did it rescue any dental defects of Dmp1−/− mice. Furthermore, the ARHR patient with a M1V mutation developed a typical rachitic dental defect that is similar to those of other ARHR patients.3,25
Interestingly, our recent findings suggest that there might be a nuclear isoform of DMP1 (referred to as ‘nuDMP1’) translated from an alternative downstream in-frame start codon (AUG210 or AUG227 for mouse Dmp1) of the same mRNA that encodes the secretory DMP1.28,29 This nuDMP1 form lacks the ER-entry signal sequence and the 37 kDa NH2-terminal sequence, but maintains the NLS sequence in its COOH-terminal. Our studies revealed that the nuDMP1 enters the nucleus and regulates cell differentiation in vitro.28,29 It is possible that the 37 kDa NH2-terminal sequence may present a steric hindrance that interferes with the function of nuDMP1, as the 57 kDa COOH-terminal fragment of Dmp1 transgene fully rescued the Dmp1−/− defects.23 However, the intact DMP1 form with the mutated cleavage sites (i.e., no 37 kDa NH2 is removed from the COOH-terminal fragment) failed to do so.30,31 Currently, we plan to test this theory by crossing the nuDMP1 transgene into the Dmp1−/− background.
In summary, we have demonstrated that the nucleus-targeted full-length DMP1 failed to rescue the dental defects in Dmp1−/− mice, suggesting that full-length DMP1 plays no apparent role in the nucleus in vivo during odontogenesis as is widely believed.
References
Fisher LW, Torchia DA, Fohr B et al. Flexible structures of SIBLING proteins, bone sialoprotein, and osteopontin. Biochem Biophys Res Commun 2001; 280( 2): 460–465.
Feng JQ, Ward LM, Liu S et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet 2006; 38( 11): 1310–1315.
Jiang B, Cao Z, Lu Y et al. DMP1 C-terminal mutant mice recapture the human ARHR tooth phenotype. J Bone Miner Res 2010; 25( 10): 2155–2164.
Lu Y, Ye L, Yu S et al. Rescue of odontogenesis in Dmp1-deficient mice by targeted re-expression of DMP1 reveals roles for DMP1 in early odontogenesis and dentin apposition in vivo. Dev Biol 2007; 303( 1): 191–201.
Ye L, MacDougall M, Zhang S et al. Deletion of dentin matrix protein-1 leads to a partial failure of maturation of predentin into dentin, hypomineralization, and expanded cavities of pulp and root canal during postnatal tooth development. J Biol Chem 2004; 279( 18): 19141–19148.
Ye L, Zhang S, Ke H et al. Periodontal breakdown in the Dmp1 null mouse model of hypophosphatemic rickets. J Dent Res 2008; 87( 7): 624–629.
George A, Sabsay B, Simonian PA et al. Characterization of a novel dentin matrix acidic phosphoprotein. Implications for induction of biomineralization. J Biol Chem 1993; 268( 17): 12624–12630.
Feng JQ, Huang H, Lu Y et al. The dentin matrix protein 1 (Dmp1) is specifically expressed in mineralized, but not soft, tissues during development. J Dent Res 2003; 82( 10): 776–780.
Fen JQ, Zhang J, Dallas SL et al. Dentin matrix protein 1, a target molecule for Cbfa1 in bone, is a unique bone marker gene. J Bone Miner Res 2002; 17( 10): 1822–1831.
He G, Dahl T, Veis A et al. Nucleation of apatite crystals in vitro by self-assembled dentin matrix protein 1. Nat Mater 2003; 2( 8): 552–558.
Gajjeraman S, Narayanan K, Hao J et al. Matrix macromolecules in hard tissues control the nucleation and hierarchical assembly of hydroxyapatite. J Biol Chem 2007; 282( 2): 1193–1204.
Gericke A, Qin C, Sun Y et al. Different forms of DMP1 play distinct roles in mineralization. J Dent Res 2010; 89( 4): 355–359.
Tartaix PH, Doulaverakis M, George A et al. In vitro effects of dentin matrix protein-1 on hydroxyapatite formation provide insights into in vivo functions. J Biol Chem 2004; 279( 18): 18115–18120.
Wu H, Teng PN, Jayaraman T et al. Dentin matrix protein 1 (DMP1) signals via cell surface integrin. J Biol Chem 2011; 286( 34): 29462–29469.
Eapen A, Ramachandran A, Pratap J et al. Activation of the ERK1/2 mitogen-activated protein kinase cascade by dentin matrix protein 1 promotes osteoblast differentiation. Cells Tissues Organs 2011; 194( 2/3/4): 255–260.
Narayanan K, Ramachandran A, Hao J et al. Dual functional roles of dentin matrix protein 1. Implications in biomineralization and gene transcription by activation of intracellular Ca2+ store. J Biol Chem 2003; 278( 19): 17500–17508.
Ravindran S, Narayanan K, Eapen AS et al. Endoplasmic reticulum chaperone protein GRP-78 mediates endocytosis of dentin matrix protein 1. J Biol Chem 2008; 283( 44): 29658–29670.
Braut A, Kalajzic I, Kalajzic Z et al. Col1α1-GFP transgene expression in developing incisors. Connect Tissue Res 2002; 43( 2/3): 216–219.
Qin C, Brunn JC, Cook RG et al. Evidence for the proteolytic processing of dentin matrix protein 1. Identification and characterization of processed fragments and cleavage sites. J Biol Chem 2003; 278( 36): 34700–34708.
Moses KD, Butler WT, Qin C . Immunohistochemical study of small integrin-binding ligand, N-linked glycoproteins in reactionary dentin of rat molars at different ages. Eur J Oral Sci 2006; 114( 3): 216–222.
Wang X, Wang S, Lu Y et al. FAM20C plays an essential role in the formation of murine teeth. J Biol Chem 2012; 287( 43): 35934–35942.
Fisher LW, Stubbs JT 3rd, Young MF . Antisera and cDNA probes to human and certain animal model bone matrix noncollagenous proteins. Acta Orthop Scand Suppl 1995; 266: 61–65.
Lu Y, Yuan B, Qin C et al. The biological function of DMP-1 in osteocyte maturation is mediated by its 57-kDa C-terminal fragment. J Bone Miner Res 2011; 26( 2): 331–340.
Farrow EG, Davis SI, Ward LM et al. Molecular analysis of DMP1 mutants causing autosomal recessive hypophosphatemic rickets. Bone 2009; 44( 2): 287–294.
Turan S, Aydin C, Bereket A et al. Identification of a novel dentin matrix protein-1 (DMP-1) mutation and dental anomalies in a kindred with autosomal recessive hypophosphatemia. Bone 2010; 46( 2): 402–409.
Prasad M, Butler WT, Qin C . Dentin sialophosphoprotein in biomineralization. Connect Tissue Res 2010; 51( 5): 404–417.
Zhu Q, Gibson MP, Liu Q et al. Proteolytic processing of dentin sialophosphoprotein (DSPP) is essential to dentinogenesis. J Biol Chem 2012; 287( 36): 30426–30435.
Wang S, Huang Y, Qin C et al. Nuclear DMP1 (nuDMP1) is generated by alternative initiation of translation. Seattle, WA: IADR, 2013. Available at https://iadr.confex.com/iadr/13iags/webprogram/Paper173877.html (accessed March 17 2013).
Siyam A, Wang S, Qin C et al. Nuclear localization of DMP1 proteins suggests a role in intracellular signaling. Biochem Biophys Res Commun 2012; 424( 3): 641–646.
Sun Y, Prasad M, Gao T et al. Failure to process dentin matrix protein 1 (DMP1) into fragments leads to its loss of function in osteogenesis. J Biol Chem 2010; 285( 41): 31713–31722.
Sun Y, Lu Y, Chen L et al. DMP1 processing is essential to dentin and jaw formation. J Dent Res 2011; 90( 5): 619–624.
Acknowledgements
We are grateful to Jeanne Santa Cruz for her assistance with the editing of this article. We thank Professor Kream and Professor Lichtler (University of Connecticut Health Center, Farmington, CT, USA) for graciously providing the mammalian expression vector; and Professor Larry Fisher (NIDCR, National Institutes of Health, Bethesda, MD, USA) for generously providing anti-Biglycan monoclonal antibody. This study was supported by NIH grants DE018486 and R56 DE022789 to Jian-Quan Feng, DE023365 to Yong-Bo Lu and a scholarship from the Chinese State Scholarship Fund to Shu-Xian Lin (2010627108).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Note: Supplementary Information for this article can be found on International Journal of Oral Science's website .
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Lin, SX., Zhang, Q., Zhang, H. et al. Nucleus-targeted Dmp1 transgene fails to rescue dental defects in Dmp1 null mice. Int J Oral Sci 6, 133–141 (2014). https://doi.org/10.1038/ijos.2014.44
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijos.2014.44
