
Abstract
Background:
Ghrelin is the only orexigenic hormone known to stimulate food intake and promote obesity and insulin resistance. We recently showed that plasma ghrelin is controlled by butyrylcholinesterase (BChE), which has a strong impact on feeding and weight gain. BChE knockout (KO) mice are prone to obesity on high-fat diet, but hepatic BChE gene transfer rescues normal food intake and obesity resistance. However, these mice lack brain BChE and still develop hyperinsulinemia and insulin resistance, suggesting essential interactions between BChE and ghrelin within the brain.
Methods:
To test the hypothesis we used four experimental groups: (1) untreated wild-type mice, (2) BChE KO mice with LUC delivered by adeno-associated virus (AAV) in combined intravenous (i.v.) and intracerebral (i.c.) injections, (3) KO mice given AAV for mouse BChE (i.v. only) and (4) KO mice given the same vector both i.v. and i.c. All mice ate a 45% calorie high-fat diet from the age of 1 month. Body weight, body composition, daily caloric intake and serum parameters were monitored throughout, and glucose tolerance and insulin tolerance tests were performed at intervals.
Results:
Circulating ghrelin levels dropped substantially in the KO mice after i.v. AAV–BChE delivery, which led to normal food intake and healthy body weight. BChE KO mice that received AAV–BChE through i.v. and i.c. combined treatments not only resisted weight gain on high-fat diet but also retained normal glucose and insulin tolerance.
Conclusions:
These data indicate a central role for BChE in regulating both insulin and glucose homeostasis. BChE gene transfer could be a useful therapy for complications linked to diet-induced obesity and insulin resistance.
Similar content being viewed by others


Introduction
Butyrylcholinesterase (BChE, EC 3.1.1.8) is present in all mammals, synthesized by the liver and secreted into the circulation. BChE is abundant in the bloodstream, but is also widespread elsewhere in the body, including the brain, skin, muscles, intestine, lung, stomach, spleen, kidney, heart, thyroid and spinal cord.1 BChE previously attracted attention as a bioscavenger of drugs and organophosphate or carbamate insecticides.2 Recent findings point toward a more specific physiological role in hydrolyzing the appetite-promoting hormone, 'ghrelin'.3, 4 Ghrelin is an acylated peptide released mostly by the stomach, but it crosses the blood–brain barrier to communicate with central rewarding systems.5 BChE modulates ghrelin's biological functions by cleaving the peptide's n-octanoyl group, leaving an inactive desacyl peptide.6, 7, 8 Thus, BChE orchestrates many physiological pathways involving ghrelin.
Large clinical studies have reported that low serum BChE serves as a biochemical marker in clinical conditions involving hepatocellular impairment from acute and chronic liver damage, cirrhosis and liver metastases.9 Low plasma BChE levels are also found in protein–energy malnutrition and inflammatory states.10 On the contrary, plasma BChE activity is elevated in obese subjects and patients with type 1 or type 2 diabetes.10, 11 High plasma BChE activity is associated with aberrant lipid profiles, insulin resistance and hypertension.11, 12 Together, these findings suggest a role for BChE in many metabolic functions and, possibly, in the insulin resistance syndrome. However, it is not known whether high BChE levels are a cause of such metabolic lesions, or whether they are a response to them.
In light of BChE's impact on ghrelin regulation and its altered expression in obese and diabetic patients, we hypothesize that it serves to adjust energy balance, especially on rich diets. To investigate physiological roles of BChE, 10 years ago, Lockridge and co-workers13 generated the first BChE knockout (KO) mice. Their only observed phenotype was readily growing obese on high-fat diets.14 We recently confirmed that result while also revealing that their plasma ghrelin was 50% higher.15 However, this effect was completely suppressed by one systemic injection of adeno-associated viral vector coding for BChE. Surprisingly, however, although treated KO mice retained healthy weight and normal calorie intake, they still developed hyperinsulinemia.15 A plausible explanation is that systemic BChE vector injection elevates enzyme only in the periphery, leaving nonexistent BChE in the brain. Here, we used bilateral vector brain injections to dissect the functional roles of peripheral and central BChE in regulating body fat as well as insulin secretion.
Materials and methods
Animal subjects and ethics
Adult male wild-type C57BL/6 and BChE KO mice of the same strain were obtained from Jackson Labs (Bar Harbor, ME, USA) under protocol A53015 approved by Mayo Clinic's Institutional Animal Care and Use Committee. Experiments were conducted in accord with the Guide for Care and Use of Laboratory Animals16 in a facility approved by the American Association for the Accreditation of Laboratory Animal Care. Under our protocol, 1-month-old mice with initially similar body weight were fed for 6 months on a diet containing 45% calories from fat, 20% from protein and 35% from carbohydrate (Research Diet).
Intravenous injection
Complementary DNA encoding luciferase (Luc) or mouse BChE was subcloned into an adeno-associated virus (AAV) backbone. The resulting transfer vectors were co-transfected into HEK293T cells with helper vectors pHELP (Applied Viromics, Fremont, CA, USA) and pAAV 2/8 (University of Pennsylvania) as described.17 Virus in cell lysates was isolated by ultracentrifugation, and viral particles were quantitated by real-time PCR. Vector (200 μl, 1 × 1012 viral particles) was given to 10-week-old mice via tail-vein, followed by 200 μl of 0.9% sterile NaCl.
Intracerebral injection
BChE KO mice (10-week-old) were anesthetized with 50 mg kg−1 pentobarbital, and 10 μl of AAV–BChE containing 1.5 × 1012 viral particles was delivered (1 μl min−1) in two injections, bilaterally targeting the hypothalamic arcuate nucleus. Intracerebral injection was performed on a Kopf stereotaxic frame with mouse adapters at coordinates: A=2.1 mm, L=0.5 mm, V=5.0 MM. Cannulae were left in place for 3 min and then withdrawn.
Body composition and food intake
Fat mass and lean mass were assessed in an EchoMRI-100 body composition analyzer (EchoMRI LLC, Houston, TX, USA). Food intake was determined daily for 2 weeks in singly housed mice with cage bedding replaced by 'Iso-pads' in order to account for every grain of residual food.
Plasma analysis
Random-fed samplings were made between 0900 and 1000 hours, and fasting samples were obtained at 1400 hours after 6 h of food deprivation. Blood glucose concentrations were measured by blood glucometers from mouse tail tips (Bayer HealthCare, Mishawaka, IN, USA). Other parameters were measured from plasma. Blood (~0.2 ml) was taken by cheek puncture with a mouse-lancet in ice-cooled EDTA tubes and centrifuged for 10 min at 8000 g. Insulin concentrations were measured by an ELISA kit (Alpco, Salem, NH, USA). BChE activity was determined by Ellman assay as described previously.18 Ghrelin samples were taken after 16 h fasting and collected in cooled EDTA-treated tubes with 0.1 vol of 1N HCl, protected by proteinase inhibitors (1 mM p-hydroxymercuribenzoic acid; Sigma-Aldrich, St Louis, MO, USA and 1.5 μM aprotinin; Roche). Samples were centrifuged 10 min at 8000 g and stored at −80 °C. ELISA kits were used to measure ghrelin and desacyl-ghrelin levels (Cayman Chemical, Ann Arbor, MI, USA).
Glucose tolerance test and insulin tolerance test
Six-hour-fasted mice were given 1.0 g kg−1 of glucose (for glucose tolerance test) or 0.75 U kg−1 of Eli Lilly human insulin (for insulin tolerance test) by intraperitoneal injection. Blood glucose concentration was monitored before and 15, 30, 60, 90 and 120 min after injection. Mice were single-caged in random order.
Histochemical BChE detection
Cryostat brain sections (14 μm) were fixed in 4% paraformaldehyde (pH 7.4) for 30 min. BChE activity was examined by Koelle–Friedenwald histochemistry with minor modification.19 Brain sections were incubated overnight in a copper glycinate medium (pH 5.0) containing 3 mM copper sulfate, 10 mM glycine, 35 mM sodium acetate, 3 mM butyrylthiocholine and 200 μM 1,5-bis(4-allyldimethylammoniumphenyl) pentan-3-one dibromide (BW284C51; Sigma-Aldrich). After four cycles of rinsing with distilled water, slices were incubated in 160 mM sodium sulfide buffer (pH 7.5) for 30 min, followed by another four water rinses. The reaction was finished up in 1% silver nitrate for 30 min. Rinsed sections were air-dried and mounted in EZ-Mount mounting medium (Thermo Scientific, Waltham, MA, USA). Images were captured on a light microscopic (Carl Zeiss Microscopy GmbH, Jena, Germany).
Data analysis
Two-group comparisons were conducted with a two-tailed t-test, and P<0.05 was considered significant. Daily food intake data were analyzed by two-factor mixed analysis of variance followed by Holm–Sidak multiple comparison tests.20 Sample sizes (10 mice per group) were chosen in light of our previous experiments of similar design and supported by previous power analysis. The variation of measured responses was similar within treatment groups.
Results
BChE expression in vector-treated KO mice
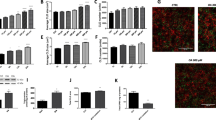
As we and others have consistently observed excess body weight in BChE KO male mice on high-fat diets, we hypothesized that both central and peripheral BChE must have an impact on fat metabolism. To test this idea, we treated BChE KO mice with AAV vector for mouse BChE in two ways: (1) via tail-vein (intravenous, i.v.), which led to high levels of circulating BChE and (2) double injections (i.v. and intracerebral (i.c.)), which generated abundant BChE in both peripheral and central tissues (Table 1 and Figure 1). A rich diet (45% calorie fat) was provided to age-matched wild-type and BChE KO mice from 4 weeks of age. Vectors were injected when the mice reached 10 weeks. Vector carrying the Luc gene served as another treatment control. Sham injection with i.c. saline was carried out in the single i.v. injection group. Giving BChE vector to KO mice led to stably high enzyme expression in plasma (>50 U ml−1), well above natural levels in wild-type controls (~1.6 U ml−1, Table 1). Accompanying the high BChE expression was a large drop in plasma acyl ghrelin, and a comparable rise in desacyl-ghrelin, altering the ratio between the two peptides (Table 1). Activity staining of coronal brain sections revealed widespread BChE expression in most brain areas, with highest levels in the triangular septal nucleus, striatum, thalamus and hypothalamus, but much lower levels in the cortex (Figure 1).

BChE activity in brains of wild-type and BChE KO mice with and without BChE gene transfer. (a) Overview of BChE activity in coronal brain sections from 7-month-old mice with the indicated treatments given 5 months previously. Note that even wild-type mice express very moderate BChE activity. (b) Higher-magnification images of cortex, triangular septal nucleus, striatum, thalamus and hypothalamus from the double injections of AAV–BChE from i.v. and i.c. Ts, triangular septal nucleus; SFO, subfornical organ; CPu, caudate putamen; GP, globus pallidus; PVA, paraventribular thalamic nucleus; PT, paratenial thalamic nucleus; VMHC, ventromedial hypothalamic nucleus; ARC, arcuate hypothalamic nucleus.
Body composition and food intake
We assessed the effect of vector treatments on body mass and composition over time. In the first week after vector injections, the mice in all groups showed similar and minor weight losses. This effect, likely owing to surgical stress, dissipated within 6 days (Figure 2a). After 18 weeks, however, the KOs given no BChE vector were 45% heavier than those at the outset. In contrast, over the same period, weight gain was only 20% in the mice given BChE vector, i.v., or i.v.+i.c., (Figure 2b). The excess weight gain in mice with no BChE was almost exclusively due to an abnormal increase in fat mass, which was 60% greater than in wild-type controls and BChE vector-treated KO mice (Figure 2c). Terminal tissue harvests revealed that inguinal but not epididymal fat pads were substantially heavier in untreated KO mice than in wild type, but they were equivalent to the normal controls in the vector-treated KO mice (Figure 2d).

Effect of high-fat diet on weight and body composition in wild-type and BChE KO mice fed a 45-calorie high-fat diet from the age of 4 weeks and given vectors at 10 weeks. (a) Time course of body weight in mice transduced with BChE or AAV-Luc by single i.v. injection or combined i.v.–i.c. injections. (b) Body weights at time of necropsy (7 months). (c) Terminal fat and lean mass across entire body. (d) Terminal fat mass in key adipose tissues. Results are means±s.e.m. (n=10 per group). **P<0.01, ***P<0.001 versus wild-type control; ###P<0.001 versus BChE KO treated with AAV-Luc control.
Careful assessment of daily food intake throughout the 2-week study confirmed that untreated KO mice ate more food than wild-type controls and vector-treated KO mice (Figure 3a). Average weekly caloric intake in the KO group was ~10% more than the other three groups (Figure 3b). Double vector injections (i.c.+i.v.) had no added effect on energy consumption compared with single i.v. injections.

Daily caloric intake in single-housed mice. (a) Caloric intake of individual 5-month-old mice was measured every day for 14 days. (b) Cumulative food intake for 1 week. Results are means±s.e.m. (n=10 per group). **P<0.01, ***P<0.001 versus wild-type control; #P<0.05 versus BChE KO treated with AAV-Luc control; n.s., not significant.
Glucose homeostasis
Human obesity is strongly associated with type 2 diabetes, but despite the aberrant body weights in our fat mice, their fasting blood glucose remained closely similar to those in wild-type and vector-treated KO mice (Figure 4a). However, when we challenged fasted animals with intraperitoneal glucose (1.0 g kg−1), the KO mice given double BChE vector treatments (i.v. and i.c.) cleared injected sugar faster than singly treated cohorts (i.v. only, Figure 4b). This effect was reflected in an ~20% reduction in 'area under the curve' relative to wild-type control and untreated KO (Figure 4c). In contrast, KO mice singly treated with i.v. AAV–BChE vector exhibited no differences in glucose tolerance as compared to the KO and wild-type mice that received no vector (Figures 4b and c). Because insulin strongly affects blood glucose, we followed its levels during the glucose tolerance test. After glucose administration, insulin rose noticeably in the untreated and i.v vector-treated KO mice after glucose administration but not in the double i.v. and i.c. treated KO (Figure 4d).

Blood glucose and glucose tolerance. (a) Blood glucose in 6-month-old mice on random feeding (measured at 1000 hours) or after 6 h of fasting (1400 hours). (b–d) Blood glucose levels, area under the curve (AUC) and blood insulin levels during glucose tolerance testing (intraperitoneal (i.p.) 1.0 g kg−1 glucose) when mice were 5 months of age. **P<0.01 versus wild-type control; #P<0.05 versus BChE KO treated with AAV-Luc control.
Further study showed that BChE KO mice developed a hyperinsulinemia that persisted in both fed and fasting status (Figure 5a). Likewise, KO mice given only i.v. AAV–BChE treatment also showed elevated insulin levels (Figure 5a) and were less sensitive than wild-type controls in the insulin tolerance test (Figures 5b and c). This outcome implies that, despite healthy body weight and normal food intake, our singly treated mice still became diabetic. In contrast, doubly treated mice (i.v.+i.c.) retained fed and fasting insulin levels comparable to wild-type cohorts, and were also similar in their response to insulin (Figures 5b and c).

Insulin levels and insulin tolerance. (a) Plasma insulin in 6-month-old random-fed animals was measured at 1000 hours or after 6 h fasting (1400 hours). (b, c) Blood glucose response to bolus insulin (0.75 U kg−1, i.p.) and corresponding area under the curve. *P<0.05 versus wild-type control; **P<0.01 versus wild-type control; #P<0.05, ##P<0.01 versus BChE KO treated with AAV-Luc control.
Discussion
Our study showed that diet-induced metabolic lesions in BChE KO mice were fully rescued by simultaneous enzyme overexpression in the brain as well as in the bloodstream. The outcomes of systemic vector alone contrasted strikingly with dual treatment with systemic and cerebral vector. We think this result points to an unexpected role for brain BChE. Thus, in systemically treated KO mice, high circulating BChE is able to bring down plasma ghrelin levels and suppress overweight and overeating. Nevertheless, these healthy lean KO mice that lacked brain BChE still developed diabetic symptoms of hyperinsulinemia and impaired insulin sensitivity. In contrast, coupling systemic with i.c. injection provided widespread BChE expression in the brain and erased all obesity-induced perturbations in glucose and insulin tolerance.
Body mass differences in this study were almost exclusively due to altered fat mass. This outcome aligns well with our previous report that BChE has a sparing effect on lean mass.15 That effect likely reflected altered ghrelin signaling in peripheral and central systems. Ghrelin originates largely from the stomach, but it signals to the brain through multiple pathways.21 Ghrelin is also produced in certain brain regions, including the hypothalamus, and it activates NPY/AgRP neurons that stimulate feeding and decrease energy expenditure.22, 23 The hypothalamus is critical in relaying afferent signals from the gut and brainstem and in processing efferent signals that modulate food intake and energy expenditure.24, 25 There are three possible pathways for BChE to influence ghrelin's appetite-inducing effect. First, circulating BChE can hydrolyze ghrelin in the bloodstream before it crosses the blood–brain barrier. Second, central BChE can regulate ghrelin produced locally in the hypothalamus where it may directly affect various hypothalamic nuclei. Third, BChE in the stomach may reduce the ghrelin signaling that reaches the brain via the afferent vagal nerve to the central motor vagus nuclei.
High-fat feeding leads to weight gain and obesity26 and is also associated with insulin resistance.27 Although our glucose tolerance and insulin tolerance tests showed impaired glucose clearance and reduced insulin sensitivity in BChE KO mice, blood glucose levels were essentially normal. As in many other diabetic models,28 impaired insulin sensitivity in the BChE KO mice was associated with increased insulin levels and secretion in response to a glucose challenge. We saw much better glucose homeostasis in the bilaterally treated KO mice, evidenced by increased glucose disposal, decreased insulin secretion and increased sensitivity to insulin injection. The improved insulin responsiveness in vector-treated BChE KO mice may be due to the altered central ghrelin levels and the sensitivity of its receptor. Ghrelin inhibits insulin release in mice, rat and humans.29 Ablation of ghrelin or its receptor enhances insulin release and prevents impaired glucose tolerance in high-fat diet-induced obese mice.30, 31 Ghrelin removal also improves diabetic symptoms in genetically leptin-deficient mice.32, 33 Overexpressing BChE in the brain, especially in the hypothalamus, may re-sensitize ghrelin's receptor and thereby maximize glucose-induced insulin release, enabling insulin secretion to meet the high demand associated with diet-induced obesity.
Neuronal pathways link the central nervous system with the autonomic innervation of the endocrine pancreas, which controls glucose homeostasis.34 Cholinergic pathways have an important role in maintaining energy and glucose homeostasis.35, 36 The cholinergic vagus nerve has been shown to stimulate insulin secretion and pancreatic cell proliferation.37, 38 It is plausible that, under a long-term high-fat diet, BChE, despite low efficiency in hydrolyzing acetylcholine, may modulate the cholinergic pathway for insulin secretion and consequently aid glucose homeostasis.
Our studies suggest that elevating BChE expression with AAV vector will have beneficial effects in controlling food intake and obesity-related disorders, and may be of future clinical value. A large excess of BChE in the blood and brain caused no obvious deficiency of cognition or motor function in our mice. In fact, for other reasons we have conducted extensive toxicology studies on the same viral vector without finding any physiologic effect from BChE enzyme or its viral host in healthy mice.39 For that reason, a role for BChE in regulating insulin sensitivity was unexpected and has not yet been rigorously defined. However, this role may well be to serve in modulating ghrelin levels and ghrelin receptor sensitivity to affect lipid metabolism. If so, BChE expression vectors may prove effective as therapeutics for obesity, type 2 diabetes and related conditions.
The present study has provided unexpected evidence that BChE in the central brain may have important roles in clearing blood glucose and modulating insulin sensitivity. We recognize a few limitations of our work. One is that different viral vector doses were used for the i.v. and i.c. treatments that led to altered plasma levels of BChE and ghrelin. Second, the specific areas of the brain crucial to BChE’s central role remain to be pinpointed, although the hypothalamus and dorsal motor nuclei of the vagus nerve are likely involved. Third, the role of ghrelin and its receptor sensitivity in the BChE KO mice model is worth pursuing with genetic approaches. Fourth, new viral vectors with improved CNS access need study to obviate the invasiveness of intracranial vector delivery. Finally, it should be of great interest to pinpoint other pathways by which BChE may have an impact on diet-induced obesity.
References
Lockridge O . Review of human butyrylcholinesterase structure, function, genetic variants, history of use in the clinic, and potential therapeutic uses. Pharmacol Ther 2015; 148: 34–46.
Marsillach J, Costa LG, Furlong CE . Protein adducts as biomarkers of exposure to organophosphorus compounds. Toxicology 2013; 307: 46–54.
De Vriese C, Gregoire F, Lema-Kisoka R, Waelbroeck M, Robberecht P, Delporte C . Ghrelin degradation by serum and tissue homogenates: identification of the cleavage sites. Endocrinology 2004; 145: 4997–5005.
Chen VP, Gao Y, Geng L, Parks RJ, Pang YP, Brimijoin S . Plasma butyrylcholinesterase regulates ghrelin to control aggression. Proc Natl Acad Sci USA 2015; 112: 2251–2256.
Kojima M, Kangawa K . Ghrelin: structure and function. Physiol Rev 2005; 85: 495–522.
Schopfer LM, Lockridge O, Brimijoin S . Pure human butyrylcholinesterase hydrolyzes octanoyl ghrelin to desacyl ghrelin. Gen Comp Endocrinol 2015; 224: 61–68.
Brimijoin S, Chen VP, Pang Y-P, Geng L, Gao Y . Physiological roles for butyrylcholinesterase: a BChE-ghrelin axis. Chem-Biol Interact 2016; 259: 271–275.
Chen VP, Gao Y, Geng L, Brimijoin S . Radiometric assay of ghrelin hydrolase activity and 3H-ghrelin distribution into mouse tissues. Biochem Pharmacol 2015; 98: 732–739.
Santarpia L, Grandone I, Contaldo F, Pasanisi F . Butyrylcholinesterase as a prognostic marker: a review of the literature. J Cachexia Sarcopenia Muscle 2013; 4: 31–39.
Lampon N, Hermida-Cadahia EF, Riveiro A, Tutor JC . Association between butyrylcholinesterase activity and low-grade systemic inflammation. Ann Hepatol 2012; 11: 356–363.
Randell E, Mathews M, Zhang H, Seraj J, Sun G . Relationship between serum butyrylcholinesterase and the metabolic syndrome. Clin Biochem 2005; 38: 799–805.
Sridhar GR, Rao AA, Srinivas K, Nirmala G, Lakshmi G, Suryanarayna D et al. Butyrylcholinesterase in metabolic syndrome. Med Hypotheses 2010; 75: 648–651.
Li B, Duysen EG, Saunders TL, Lockridge O . Production of the butyrylcholinesterase knockout mouse. J Mol Neurosci 2006; 30: 193–195.
Li B, Duysen EG, Lockridge O . The butyrylcholinesterase knockout mouse is obese on a high-fat diet. Chem Biol Interact 2008; 175: 88–91.
Chen VP, Gao Y, Geng L, Stout MB, Jensen MD, Brimijoin S . Butyrylcholinesterase deficiency promotes adipose tissue growth and hepatic lipid accumulation in male mice on high-fat diet. Endocrinology 2016; 157: 3086–3095.
National Research Council. Guide for the Care and Use of Laboratory Animals, 8th edn. National Academies Press: Washington, DC, USA, 2011..
Geng L, Gao Y, Chen X, Hou S, Zhan C-G, Radic Z et al. Gene transfer of mutant mouse cholinesterase provides high lifetime expression and reduced cocaine responses with no evident toxicity. PLoS One 2013; 8: e67446.
Chen VP, Xie HQ, Chan WK, Leung KW, Chan GK, Choi RC et al. The PRiMA-linked cholinesterase tetramers are assembled from homodimers: hybrid molecules composed of acetylcholinesterase and butyrylcholinesterase dimers are up-regulated during development of chicken brain. J Biol Chem 2010; 285: 27265–27278.
Hammond PI, Jelacic T, Padilla S, Brimijoin S . Quantitative, video-based histochemistry to measure regional effects of anticholinesterase pesticides in rat brain. Anal Biochem 1996; 241: 82–92.
Abdi H. The Bonferonni and Šidák corrections for multiple comparisons. In: Salkind NJ (ed). Encyclopedia of Measurement and Statistics, Sage: Thousand Oaks, CA, USA, 2007, pp 103–107..
Banks WA, Tschop M, Robinson SM, Heiman ML . Extent and direction of ghrelin transport across the blood-brain barrier is determined by its unique primary structure. J Pharmacol Exp Ther 2002; 302: 822–827.
Cowley MA, Smith RG, Diano S, Tschop M, Pronchuk N, Grove KL et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 2003; 37: 649–661.
Korbonits M, Goldstone AP, Gueorguiev M, Grossman AB . Ghrelin–a hormone with multiple functions. Front Neuroendocrinol 2004; 25: 27–68.
Simpson KA, Martin NM, Bloom SR . Hypothalamic regulation of food intake and clinical therapeutic applications. Arq Bras Endocrinol Metabol 2009; 53: 120–128.
Palkovits M . Hypothalamic regulation of food intake. Ideggyogy Sz 2003; 56: 288–302.
Jen KL . Effects of diet composition on food intake and carcass composition in rats. Physiol Behav 1988; 42: 551–556.
Storlien LH, James DE, Burleigh KM, Chisholm DJ, Kraegen EW . Fat feeding causes widespread in vivo insulin resistance, decreased energy expenditure, and obesity in rats. Am J Physiol 1986; 251 (5 Pt 1): E576–E583.
Shanik MH, Xu Y, Skrha J, Dankner R, Zick Y, Roth J . Insulin resistance and hyperinsulinemia: is hyperinsulinemia the cart or the horse? Diabetes Care 2008; 31 (Suppl 2): S262–S268.
Dezaki K . Ghrelin function in insulin release and glucose metabolism. Endocr Dev 2013; 25: 135–143.
Dezaki K, Sone H, Koizumi M, Nakata M, Kakei M, Nagai H et al. Blockade of pancreatic islet-derived ghrelin enhances insulin secretion to prevent high-fat diet-induced glucose intolerance. Diabetes 2006; 55: 3486–3493.
Lin L, Saha PK, Ma X, Henshaw IO, Shao L, Chang BH et al. Ablation of ghrelin receptor reduces adiposity and improves insulin sensitivity during aging by regulating fat metabolism in white and brown adipose tissues. Aging Cell 2011; 10: 996–1010.
Sun Y, Asnicar M, Saha PK, Chan L, Smith RG . Ablation of ghrelin improves the diabetic but not obese phenotype of ob/ob mice. Cell Metabol 2006; 3: 379–386.
Yada T, Dezaki K, Sone H, Koizumi M, Damdindorj B, Nakata M et al. Ghrelin regulates insulin release and glycemia: physiological role and therapeutic potential. Curr Diabetes Rev 2008; 4: 18–23.
Thorens B . Central control of glucose homeostasis: the brain–endocrine pancreas axis. Diabetes Metab 2010; 36 (Suppl 3): S45–S49.
Berthoud HR . The vagus nerve, food intake and obesity. Regul Peptides 2008; 149: 15–25.
Dockray GJ . The versatility of the vagus. Physiol Behav 2009; 97: 531–536.
Kiba T . Relationships between the autonomic nervous system and the pancreas including regulation of regeneration and apoptosis: recent developments. Pancreas 2004; 29: e51–e58.
de Azua IR, Gautam D, Guettier J-M, Wess J . Novel insights into the function of β-cell M(3) muscarinic acetylcholine receptors: therapeutic implications. Trends Endocrinol Metab 2011; 22: 74–80.
Murthy V, Gao Y, Geng L, LeBrasseur NK, White TA, Parks RJ et al. Physiologic and metabolic safety of butyrylcholinesterase gene therapy in mice. Vaccine 2014; 32: 4155–4162.
Acknowledgements
This research was funded by grant MNP13.05 from the Minnesota Partnership for Biotechnology and Medical Genomics, by the Mayo Foundation for Medical Research and by the National Institute on Drug Abuse (Grant DP1 DA31340-01 and DA42492).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflicts of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Chen, V., Gao, Y., Geng, L. et al. Butyrylcholinesterase regulates central ghrelin signaling and has an impact on food intake and glucose homeostasis. Int J Obes 41, 1413–1419 (2017). https://doi.org/10.1038/ijo.2017.123
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijo.2017.123
This article is cited by
-
Black and non-black population: investigation of the difference in butyrylcholinesterase activity in a healthy population in Salvador, Bahia
Irish Journal of Medical Science (1971 -) (2023)
-
Molecular and cellular mechanisms underlying the hepatoprotective role of ghrelin against NAFLD progression
Journal of Physiology and Biochemistry (2023)
-
Circulating acyl and des-acyl ghrelin levels in obese adults: a systematic review and meta-analysis
Scientific Reports (2022)
-
Novel somatic variants involved in biochemical activity of pure growth hormone-secreting pituitary adenoma without GNAS variant
Scientific Reports (2021)
