
Abstract
Obesity is linked to a wide variety of cardiac changes, from subclinical diastolic dysfunction to end-stage systolic heart failure. Obesity causes changes in cardiac metabolism, which make ATP production and utilization less efficient, producing functional consequences that are linked to the increased rate of heart failure in this population. As a result of the increases in circulating fatty acids and insulin resistance that accompanies excess fat storage, several of the proteins and genes that are responsible for fatty acid uptake and metabolism are upregulated, and the metabolic machinery responsible for glucose utilization and oxidation are inhibited. The resultant increase in fatty acid metabolism, and the inherent alterations in the proteins of the electron transport chain used to create the gradient needed to drive mitochondrial ATP production, results in a decrease in efficiency of cardiac work and a relative increase in oxygen usage. These changes in cardiac mitochondrial metabolism are potential therapeutic targets for the treatment and prevention of obesity-related heart failure.
Similar content being viewed by others

Introduction
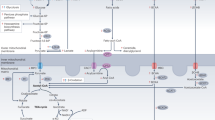
Cardiac energy metabolism is essentially a four-step process involving the following: (1) myocellular substrate uptake/selection, (2) mitochondrial ATP production and (3) ATP transfer from the site of production (mitochondrion) to (4) the site of ATP utilization (cardiac myofibril; Figure 1).1

Sequential steps in myocardial metabolism. (1) Fuel selection, (2) mitochondrial oxidation and ATP production, (3) ATP transport via CK shuttle from site of production to (4) site of utilization for contractile function.
Although glycolysis is an ATP generator, the overall ATP production is controlled largely by the rate at which the Krebs (tricarboxylic acid) cycle operates.2 Acetyl co-enzyme A (CoA), which is produced from the oxidation of fatty acids, ketone bodies, or glucose via glycolysis, and the pyruvate dehydrogenase (PDH) enzyme complex3 enters the Krebs cycle for complete oxidation. During oxidative phosphorylation, electrons, primarily obtained from oxidative metabolism of carbohydrates and fats, are transferred through the electron transport chain, a four-complex protein system embedded within the inner membrane of the mitochondria. The major function of the electron transport chain is to produce a proton electrochemical potential difference between two compartments that powers ATP synthase to generate ATP, which is used for all cardiac cellular processes.4
ATP is the heart’s only immediate source of energy for contraction, and as both systole and diastole are ATP-consuming processes,5, 6 cardiac ATP demand is very high. To keep up with this demand for continuous and efficient contraction and relaxation, the heart needs to produce around 20 times its own weight in ATP per day.1 As a result of this large energy requirement, any impairment in ATP production, transfer or utilization can have detrimental effects on cardiac function.7 Cardiac metabolism and ATP production is altered in obesity and has emerged as a candidate mechanism to explain the increase in heart failure in this population.8 Indeed, it has recently been shown that obesity, in the absence of co-morbidities, is linked to impaired myocardial high-energy phosphate metabolism9 and diastolic dysfunction,10, 11, 12 both markers of increased cardiovascular risk,13 providing a mechanistic link between altered myocardial energy production and mortality.
However, although it is well recognized that a subset of obese subjects are free of the associated metabolic co-morbidities, it is well known that the majority of obese subjects are at risk of insulin resistance, diabetes and hypertension, all of which are known to independently effect cardiac energy metabolism.5, 14 As such, isolating the effects of obesity per se on cardiac metabolism is difficult, but given the ever-increasing incidence of obesity and its links to heart failure8 and mortality,15 understanding the alterations of myocardial metabolism that occur in obesity are of great importance and may provide therapeutic options to treat or prevent cardiac dysfunction. This review focuses on the current knowledge of the changes in myocardial metabolism that occur in obesity without established co-morbidities.
Methods
Relevant articles were selected from Pubmed. The initial search term was ‘Myocardial, Metabolism, Obesity’, which revealed 1877 articles. This was refined to ‘Myocardial Energetics Obesity’ (11 articles), ‘Myocardial Substrate Selection’ (80 articles), ‘Myocardial Substrate Metabolism Obesity’ (64 articles), ‘Myocardial Substrate Metabolism Weight Loss’ (24 articles) and ‘Partial Fatty Acid Oxidation Inhibitors Heart’ (23 articles). Of these 202 articles, 88 were excluded for not having a direct relevance to obesity and 114 were finally selected for the review.
Altered myocardial substrate selection in obesity
Myocardial substrate selection is a fundamental step in myocardial metabolism. In normal heart, in the resting, fasted state, the vast majority (60–90%)16 of the acetyl CoA that enters the Krebs cycle comes from the β-oxidation of free fatty acids (FFAs),17 with 10–40% of the acetyl CoA coming from the oxidation of pyruvate, which itself is derived from either glycolysis or lactate oxidation (Figure 2).18 However, the heart is able to display great flexibility in its choice of substrate, depending on the prevailing metabolic conditions.19 For example, in the uncontrolled diabetic state, because of the combined effects of insulin resistance and high circulating FFAs, the myocardium uses fatty acids almost exclusively to support ATP synthesis.20

Normal myocardial metabolism.
This remarkable ability of the heart to switch between metabolic substrates appears to be a part of natural fetal development, where a switch in cardiac fuel preference from glucose to fatty acids occurs just after birth, when oxygen availability and dietary fat content abruptly increase, making fatty acid oxidation more preferable than glucose oxidation. The importance of this neonatal metabolic switch to fatty acid preference is apparent in children with mutations in medium-chain acyl-CoA dehydrogenase (MCAD) and very long-chain acyl-CoA dehydrogenase (VLCAD), genes involved in fatty acid β-oxidation, who develop a cardiomyopathy during periods of illness or metabolic stress.21 The importance is again highlighted in the setting of heart failure and left ventricular (LV) hypertrophy, where mitochondrial oxidative capacity is reduced and metabolism shifts back towards a reliance on glucose metabolism, resembling the fetal metabolic program.22, 23
As the heart is an extremely efficient scavenger of circulating non-esterified FFAs (up to 40% extraction fraction),24 the rate of fatty-acid uptake by the heart is primarily determined by the concentration of non-esterified fatty acids in the plasma.25 The concentration of serum FFAs is highly regulated and represents a balance between production via hormone-sensitive lipase-induced adipose tissue triglyceride breakdown and synthesis via glycerolphosphate acyltransferase.25 As hormone-sensitive lipase is activated by catecholamines and inhibited by insulin; this allows the plasma FFA concentration to rise during periods when glucose supply is limited (for example, exercise or fasting), resulting in a higher rate of cardiomyocyte uptake and utilization.25, 26
Fatty acid movement into the cardiomyocyte occurs either by passive diffusion or by protein-mediated transport across the sarcolemma via fatty acid translocase (FAT/CD36) or fatty acid-binding protein.27 Once inside the cell, control of fatty acid oxidation occurs at the level of mitochondrial uptake of fatty acids by carnitine palmitoyltransferase 1 (CPT1). CPT1 is associated with the outer mitochondrial membrane and mediates the transport of long-chain fatty acids across the membrane by binding the fatty acid moiety from acyl-CoA to long-chain acylcarnitine, which is then transported into the mitochondria.28, 29 CPT1 is inhibited by malonyl-CoA, an important regulator of fatty acid oxidation in the heart. Malonyl-CoA, the first intermediate in fatty acid synthesis, is produced by acetyl-CoA carboxylase and is broken down by malonyl-CoA decarboxylase.30 AMP-activated protein kinase regulates malonyl-CoA levels by phosphorylating and inhibiting acetyl-CoA carboxylase, increasing fatty acid oxidation (Figure 2).31 Obesity is linked to increased circulating FFA levels,32 and both human33 and animal studies34, 35 have shown increased oxidation of FFAs in obesity and insulin resistance, and a shift in substrate utilization further towards FFA metabolism (Figure 3).

Changes in myocardial metabolism in obesity (red cross denotes PPARα-mediated change). The colour reproduction of this figure is available on the International Journal of Obesity journal online.
The crucial importance of this increase in fatty acid metabolism lies in the fact that the mitochondrial redox state and, as a result, the free energy of hydrolysis of ATP are affected by the substrate oxidized. To understand this effect, we have to consider the relationship between the thermodynamic relationship, between ΔG (Gibbs free energy, a thermodynamic potential that measures the process-initiating work obtainable from a thermodynamic system) and ΔH (change in enthalpy or heat energy). Fundamentally, our body is driven by a series of controlled chemical reactions, resulting in the oxidation of carbon substrates to water and CO2. Thus, for a given amount of substance, the maximum amount of non-expansive work that can be obtained from a closed system is denoted by the Gibbs free energy. Described in 1873,36 this application of the second law of thermodynamics can be readily translated to biological systems and, in its simplest form, relates enthalpy and entropy to a conservation of energy. This is put formally as:

This equation, in part, explains why certain substrates (with higher enthalpy) yield greater potential energy to power a system; the larger the value of Gibbs free energy, the more energy that can be exchanged with the surrounding system. In non-standard chemical conditions such as those present in most biological systems,37 an alternative form of this equation is used.

This equation allows the integration of the reaction quotient (Q) into the relationship between free energy and the chemical conditions under which the reaction is taking place. In case of cellular substrate energetics, the final common endpoint for the complete oxidation of carbon fuels is the conservation of energy in the phosphate bonds of ATP. Therefore, applying this concept to the equation above, the inherent energy stored in this bond (ΔGATP hydrolysis) can be calculated from the equation38, 39, 40, 41, 42, 43, 44, 45, 46, 47

Despite the apparent simplicity of oxidizing substrates to liberate energy to perform work, the useful free energy of substrate combustion is influenced by the architecture of the metabolic pathway and the enthalpy of that particular substrate. For this reason, the available free energy to perform work, the free energy of ATP hydrolysis (ΔG′ATP hydrolysis), is not equivalent for all dietary fuels. As the conversion of ADP+Pi to ATP is driven by the electrochemical potential difference across the mitochondrial membranes, the equation for free energy can now be expressed as:

(where ΔG′ is the free energy, n is the number of electrons, F is the Faraday constant and ΔE is the difference in redox potential between ‘Inter’ and ‘Matrix’, denoting the separate mitochondrial phases partitioned by the inner mitochondrial membrane.48, 46 It then becomes apparent that the larger the electrical potential difference between mitochondrial phases created by the pumping of protons into the inter-mitochondrial space,49 the greater the potential free energy. An increase in redox energy of the respiratory chain results in an increase in the energy of the protons expelled from the mitochondria at the energy-conserving sites, which is then reflected in an increase in the energy of ATP hydrolysis. We can express the potential energy of this proton gradient as:

Therefore, the relative supply of reducing equivalents generated by the architecture of each pathway also has a significant influence on mitochondrial potential gradients, and thus the ΔG′ATP hydrolysis.43, 50, 51
Hence, although the electron transport chain is in itself a remarkably efficient series of biochemical reactions,52 the free energy of ATP hydrolysis is not identical for all substrates (Figure 4).53, 50, 51 Heat of combustion is also of inherent importance when considering the potential impact of mitochondrial substrate selection on energetic performance. Pyruvate, the end product of glycolysis, has a lower heat of combustion per C2 unit than palmitate, providing less potential energy to the electron transport chain.

The effect of varying substrate selection on the ΔG produced by the electron transport chain.
However, fatty acid metabolism, despite its large potential energy, is not able to provide greater mitochondrial redox power. The reasons for this lies in the architecture of fatty acid metabolism by β-oxidation, and the changes in mitochondrial membrane uncoupling proteins in response to persistently elevated FFAs. Only 50% of the reducing equivalents produced in the process of β-oxidation are able to donate electrons at complex I of the electron transport chain, whereas the remaining half are donated by FADH2 at the flavoprotein site further ‘downstream’ at complex II.48 This results in a reduced ATP yield and a loss of mitochondrial efficiency. The redox span of the respiratory chain is diminished during fat metabolism as the Q-couple is reduced. This decreases the potential difference between matrix and inter-mitochondrial membrane space, and therefore ΔG′ATP. Raised FFAs also increase the expression of uncoupling proteins,54 which decrease mitochondrial efficiency43 by allowing the passage of protons into the matrix via non-ATP-generating pathways. Indeed, when the heart is perfused with increasing concentrations of FFAs, this results in an additional oxygen cost of between 25 and 48% for the same work output when compared with glucose and insulin infusion.55 The loss of myocardial efficiency when metabolizing fat has been attributed to reductions in mitochondrial electron transport chain coupling, and the increased stoichiometric oxygen requirement to oxidize fat.56 As such, deleterious substrate selection may be a feature of obesity-related cardiomyopathy as it is in other myocardial diseases, intimately linking energetic performance and mortality.56, 57
In addition, positron emission topography studies have shown that in human obesity myocardial fatty acid uptake is increased and myocardial efficiency reduced (if calculated as cardiac work/oxygen usage).58 This is in keeping with the increased utilization of fatty acids for ATP production, and suggests either a decoupling of fatty acid oxidation and ATP production or futile cycling of substrates in the obese heart with energy wastage.59 Elevations in FFA levels are thought to increase mitochondrial uncoupling, and energy wastage, via increased myocardial uncoupling protein 3 expression.60, 61As diastole is more susceptible to ATP shortage than systole, this would then lead to a mechanism by which reduced high-energy phosphate levels, caused by increased mitochondrial uncoupling as a result of elevated FFA levels, may manifest as diastolic dysfunction, an almost universal finding in obesity.10, 11
This shift towards fatty acid metabolism appears to be a combination of reduced insulin-induced GLUT4 (glucose transporter type-4)-mediated glucose uptake,62, 63 suppressed glycolysis in the cytosol and reduced PDH flux in the mitochondria, reducing carbohydrate oxidation. Although the complexities of the inhibition of carbohydrate metabolism are not fully elucidated, the inhibition of glucose oxidation by fatty acids at the level of the PDH complex is universally reported, and has been termed the glucose-fatty acid or Randle cycle.64, 65, 66 Up until very recently, the vast majority of experimental data for altered substrate selection in obesity and insulin resistance were from ex vivo and in vitro studies. These studies are however limited, in that most generate steady-state, rather than real-time, information. The development of hyperpolarized 13C magnetic resonance, in which the 13C signal is amplified by >10 000-fold, provides a solution to this and has allowed real-time visualization of substrate uptake and metabolism. So far, these studies have been focused primarily on PDH activity, which, given its pivotal position in the glucose–fatty acid cycle, has allowed further insight into cardiac substrate selection67 and have again shown that in vivo real-time PDH activity is decreased in diabetic68 and high-fat diet animal models.69
Furthermore, in addition to the effects of increased fatty acid uptake and utilization on the production of the electrochemical gradient that powers ATP production, there is now evidence that there are intrinsic defects in the metabolic machinery of the electron transport chain (complexes I, III and IV) in human and animal models of obesity, with electron transport chain function and efficiency being reduced.70, 71, 72, 73, 35
As a result of the evidence that substrate selection alters myocardial efficiency, several novel therapies have been evaluated in the setting of ischemia, a situation where reducing myocardial oxygen consumption without decreasing cardiac work would be beneficial. In these settings, a shift in the proportions of ATP generated from fatty acid oxidation towards glucose oxidation would provide the heart with an efficient method to maintain a constant fuel source in the face of hypoxia. To date, several partial fatty acid oxidation inhibitors acting either via CPT-1 (Perhexiline)74 or via directly inhibiting fatty acid oxidation (Trimetazidine) have been shown to be beneficial in heart failure, ischemic heart disease and animal models of pulmonary hypertension.75, 76 However, therapies aimed at altering substrate metabolism in obesity have been limited to ischemia reperfusion models,77 and further investigation of the effects of fatty acid oxidation inhibitors are warranted in obesity.
Cardiac energetics and obesity
Heart failure, a well-documented sequelae of obesity,8 is associated with deranged cardiac energetics,1 that is, a decreased efficiency of substrate utilization to create the ATP necessary to drive cardiac contraction. This has also been demonstrated in other cardiovascular disorders such a hypertensive heart disease and diabetes.24, 25 Using 31P magnetic resonance spectroscopy, cardiac energetics can be assessed by quantifying the relative concentrations of phosphocreatine (PCr) and ATP in the myocardium to derive the PCr/ATP ratio, a sensitive index of the energetic state of the heart. In heart failure, the PCr/ATP ratio correlates with LV function26 and clinical status,27 and has been shown to be a better prognostic indicator than LV ejection fraction.28 Improving cardiac metabolism has been postulated as a novel treatment of heart failure.23, 29 It has been shown that in animal models of obesity35 and in humans with no other co-morbidity, abnormally low PCr/ATP ratios occur at rest, potentially due to, in addition to changes in substrate utilization, a loss of the total creatine pool in proportion to the loss of PCr, as occurs in many other forms of hypertrophy.1, 78, 79, 80, 81 Furthermore, this has been linked to altered cardiac diastolic function and is exacerbated during catecholamine stress.80
Mitochondrial metabolism and lipotoxicity in obesity
Cardiac mitochondria contain a DNA genome that encodes some of the proteins required for electron transport complexes I, III, and IV, and in addition, complex V. The remainder of the respiratory subunits, and all of the proteins required for substrate metabolism, are encoded by separate nuclear genes.82 It is becoming clear that in obesity, changes in both nuclear and mitochondrial transcription are present and are important in the production of the observed changes in cardiac metabolism.61
One of the key controllers of nuclear gene transcription, which regulates myocardial mitochondrial fatty acid oxidation, is the peroxisome proliferator-activated receptors (PPARs),83 which, when activated, induce perixosome proliferation. Peroxisomes have multiple metabolic roles, which include long- and very long-chain fatty acid oxidation.84 Three PPAR receptors have been identified, PPARγ, PPARδ and PPARα, all with different tissue expression. PPARα is expressed in the myocardium85 and is the primary transcriptional regulator of fat metabolism in tissues with the highest rates of fatty acid oxidation.86 Activation of PPARα in the heart increases the expression of several genes involved in fatty acid metabolism including the following: (a) cardiac myocellular fatty acid uptake (fatty acid transport protein, FAT/CD36, fatty acid-binding protein, acyl-CoA synthetase;87, 88, 89 (b) mitochondrial fatty acid uptake via CPT I;90 and (c) mitochondrial and peroxisomal fatty acid β-oxidation via medium-chain acyl-CoA dehydrogenase, long-chain acyl-CoA dehydrogenase, very long-chain acyl-CoA dehydrogenase and Acyl-CoA Oxidase, respectively (Figure 3).90
In the setting of insulin resistance, such as obesity, the heart initially adapts to increases in circulating fatty acid levels by increasing PPARα, resulting in a compensatory increase in myocardial fatty acid uptake and β-oxidation,91 which is believed to limit cardiac ectopic lipid accumulation. A further protective mechanism against ectopic cardiac fat deposition has been suggested in obese animal models, with increased cardiac expression of microsomal triglyceride transfer protein and increased formation of apolipoprotein B-containing lipoproteins, which are then secreted by cardiomyocytes.92
However, despite these initial adaptive/protective mechanisms, the potential for cardiac lipotoxicity in obesity has been described.93 Fatty acid inhibition of myocardial glucose use appears to be one important contributing factor.94, 95 Exposure of the heart to high levels of fatty acids can cause accumulation of lipids within cardiomyocytes increasing the intracellular pool of long-chain fatty acyl-CoA, which provides a fatty acid substrate for non-oxidative processes, including triacylglycerol, diacylglycerol and ceramide synthesis, which can lead to cell dysfunction, insulin resistance and, potentially, apoptotic cell death. A clear link between lipid accumulation and cardiomyopathy has now been established in several transgenic mouse models in which the rate of lipid uptake or esterification of fatty acids by the heart was increased or the capacity for oxidation of fatty acids was reduced in the mitochondria.93, 96
In addition to the PPARα-mediated increases in fatty acid oxidation, cardiac myocytes from Zucker obese rats have a larger proportion of FAT/CD3697, 98 located at the plasma membrane when compared with Zucker lean rats.99 Normal insulin-mediated translocation of FAT/CD36 is not seen in Zucker obese rats, supporting the notion that a substantial portion of the FAT/CD36 pool is permanently relocated to the sarcolemma in the heart in obesity, and that this enables triglyceride accumulation via increased fatty acid uptake.100 GLUT4 expression is also altered by excessive nutrient intake. In normal cardiac tissue, insulin causes the mobilization of GLUT4 from intracellular stores to the sarcolemma. However, in obesity and insulin resistance this process is reduced. When put together with the evidence of altered FAT/CD36 positioning, this suggests that excessive nutritional intake causes a pattern of distribution of FAT/CD36 and GLUT4, which is directed towards increased fatty acid uptake and ectopic fat deposition.100
Although there is good evidence that lipid accumulation can cause cardiac dysfunction, whether or not the accumulation of triglyceride in the heart is a purely maladaptive process contributing to cardiac dysfunction has recently come under scrutiny. There is now alternative evidence to suggest that cardiac triglyceride accumulation may be providing a protective role against fatty acid-induced lipotoxicity via limiting the accumulation of ceramides and diacylglycerols.101 However, regardless of whether ectopic lipid deposition is a maladaptive or a protective process, there is now strong evidence that myocardial steatosis promotes the development of insulin resistance, cardiac hypertrophy, impaired cardiac function and fatty acid-induced programmed cell death, and interstitial fibrosis.102
Adipokine regulation of myocardial metabolism
It is now well established that adipose tissue secretes a range of adipokines (for example, leptin, adiponectin, resistin, ghrelin, visfatin) that alter fat metabolism.103 Obesity affects the levels of these hormones, and two of these, namely leptin and adiponectin, have been shown to modulate myocardial substrate metabolism. Adiponectin is believed to act via PPARα to stimulate fatty acid metabolism, increase CPT1 activity and decrease malonyl-CoA inhibition of CPT1 activity.104, 105 However, as adiponectin is significantly lowered by obesity106 and fatty acid metabolism is increased, the full role of adiponectin in myocardial metabolism in obesity remains unknown. In contrast, leptin increases with increasing obesity,107 and has been shown to increase myocardial fatty acid metabolism and decrease myocardial glucose metabolism, in line with the observed changes seen in obesity. This increase in fatty acid oxidation occurs independent of changes in insulin signaling and PPARα transcriptional regulation, but may be attributable to increased fatty acid transport proteins on the plasma membrane.108 It has also been postulated that leptin has an important role in the prevention of cardiac lipotoxicity by confining the storage of excess lipids to adipocytes while simultaneously limiting the storage of intracellular lipids in myocytes and other non-adipocytes.109
The effects of weight loss on myocardial metabolism
As obesity is associated with increased myocardial fatty acid uptake and oxidation, lipotoxicity and decreased myocardial energetics, all known to be detrimental to cardiac function, understanding the effects of weight loss are of increasing importance. Weight-loss interventions have not only been shown to decrease myocardial FFA uptake without changing insulin-stimulated myocardial glucose uptake,110 but also to reduce myocardial fatty acid oxidation (per gram of LV), and that this decreased fatty acid oxidation is linked to decreased myocardial oxygen consumption (myocardial oxygen uptake per gram of LV).111 When put together, this strengthens the evidence that increased fatty acid uptake and oxidation in obesity is linked to decreased cardiac efficiency, and that weight loss partially reverses these effects. In addition to this, moderate dietary weight loss has been shown to significantly reduce myocardial triglyceride content112 and improve both myocardial energetics and diastolic function in obese subjects without cardiovascular risk factors.113 Weight-loss surgery has also been shown to provide early adjustments of the metabolic and neurohumoral pathways involved in energy homeostasis and reverse obesity-related hemodynamic, metabolic and cardiac dysfunction.114 Given this clear benefit of the reduction in fatty acid oxidation rates that accompany weight loss, further understanding of cardiac metabolism in obesity may lead to therapeutic options to modulate metabolism and treat cardiac dysfunction in obesity.
Conclusion
Obesity is an escalating problem and is linked to a spectrum of cardiac dysfunction from subclinical changes in diastolic function to severe systolic heart failure. There is now emerging evidence that alterations in myocardial substrate selection in obesity towards increased fatty acid oxidation and away from glucose metabolism, results in decreased contractile efficiency and may well underpin the susceptibility to contractile dysfunction in this population. The heart in obesity is also characterized by an accumulation of intracellular triglycerides and lipids that promote lipotoxicity and dysfunction. As novel imaging techniques are now providing a greater detail of this altered myocardial metabolism in vivo, potential targets for therapeutic interventions aimed at preventing and treating the cardiomyopathy of obesity via altering myocardial metabolism are likely to become a reality.
References
Neubauer S . The failing heart–an engine out of fuel. N Engl J Med 2007; 356: 1140–1151.
Stanley WC, Chandler MP . Energy metabolism in the normal and failing heart: potential for therapeutic interventions. Heart Fail Rev 2002; 7: 115–130.
Randle PJ, Garland PB, Hales CN, Newsholme EA . The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963; 1: 785–789.
Dedkova EN, Blatter LA . Measuring mitochondrial function in intact cardiac myocytes. J Mol Cell Cardiol 2012; 52: 48–61.
Ashrafian H, Frenneaux MP, Opie LH . Metabolic mechanisms in heart failure. Circulation 2007; 116: 434–448.
Opie LH . Substrate utilization and glycolysis in the heart. Cardiology 1971; 56: 2–21.
Varma N, Eberli FR, Apstein CS . Increased diastolic chamber stiffness during demand ischemia: response to quick length change differentiates rigor-activated from calcium-activated tension. Circulation 2000; 101: 2185–2192.
Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG et al. Obesity and the risk of heart failure. N Engl J Med 2002; 347: 305–313.
Rider OJ, Francis JM, Ali MK, Holloway C, Pegg T, Robson MD et al. Effects of catecholamine stress on diastolic function and myocardial energetics in obesity. Circulation 125: 1511–1519.
Otto ME, Belohlavek M, Khandheria B, Gilman G, Svatikova A, Somers V . Comparison of right and left ventricular function in obese and nonobese men. Am J Cardiol 2004; 93: 1569–1572.
Peterson LR, Waggoner AD, Schechtman KB, Meyer T, Gropler RJ, Barzilai B et al. Alterations in left ventricular structure and function in young healthy obese women: assessment by echocardiography and tissue Doppler imaging. J Am Coll Cardiol 2004; 43: 1399–1404.
Rider OJ, Francis JM, Ali MK, Petersen SE, Robinson M, Robson MD et al. Beneficial cardiovascular effects of bariatric surgical and dietary weight loss in obesity. J Am Coll Cardiol 2009; 54: 718–726.
Bhatia RS, Tu JV, Lee DS, Austin PC, Fang J, Haouzi A et al. Outcome of heart failure with preserved ejection fraction in a population-based study. N Engl J Med 2006; 355: 260–269.
An D, Rodrigues B . Role of changes in cardiac metabolism in development of diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol 2006; 291: H1489–H1506.
Adams KF, Schatzkin A, Harris TB, Kipnis V, Mouw T, Ballard-Barbash R et al. Overweight, obesity, and mortality in a large prospective cohort of persons 50 to 71 years old. N Engl J Med 2006; 355: 763–778.
Stanley WC, Lopaschuk GD, Hall JL, McCormack JG . Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res 1997; 33: 243–257.
Stanley WC, Recchia FA, Lopaschuk GD . Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 2005; 85: 1093–1129.
Wisneski JA, Stanley WC, Neese RA, Gertz EW . Effects of acute hyperglycemia on myocardial glycolytic activity in humans. J Clin Invest 1990; 85: 1648–1656.
Taegtmeyer H, Golfman L, Sharma S, Razeghi P, van Arsdall M . Linking gene expression to function: metabolic flexibility in the normal and diseased heart. Ann NY Acad Sci 2004; 1015: 202–213.
Wall SR, Lopaschuk GD . Glucose oxidation rates in fatty acid-perfused isolated working hearts from diabetic rats. Biochim Biophys Acta 1989; 1006: 97–103.
Kelly DP, Strauss AW . Inherited cardiomyopathies. N Engl J Med 1994; 330: 913–919.
Taegtmeyer H, Overturf ML . Effects of moderate hypertension on cardiac function and metabolism in the rabbit. Hypertension 1988; 11: 416–426.
Depre C, Shipley GL, Chen WH, Han QY, Doenst T, Moore ML et al. Unloaded heart in vivo replicates fetal gene expression of cardiac hypertrophy. Nat Med 1998; 4: 1269–1275.
Vyska K, Machulla HJ, Stremmel W, Fassbender D, Knapp WH, Notohamiprodjo G et al. Regional myocardial free fatty acid extraction in normal and ischemic myocardium. Circulation 1988; 78: 1218–1233.
Bing RJ, Siegel A, Ungar I, Gilbert M . Metabolism of the human heart. II. Studies on fat, ketone and amino acid metabolism. Am J Med 1954; 16: 504–515.
Lopaschuk GD, Collins-Nakai R, Olley PM, Montague TJ, McNeil G, Gayle M et al. Plasma fatty acid levels in infants and adults after myocardial ischemia. Am Heart J 1994; 128: 61–67.
van der Vusse GJ, van Bilsen M, Glatz JF . Cardiac fatty acid uptake and transport in health and disease. Cardiovasc Res 2000; 45: 279–293.
McGarry JD, Foster DW . Regulation of hepatic fatty acid oxidation and ketone body production. Annu Rev Biochem 1980; 49: 395–420.
McGarry JD, Woeltje KF, Kuwajima M, Foster DW . Regulation of ketogenesis and the renaissance of carnitine palmitoyltransferase. Diabetes Metab Rev 1989; 5: 271–284.
Dyck JR, Barr AJ, Barr RL, Kolattukudy PE, Lopaschuk GD . Characterization of cardiac malonyl-CoA decarboxylase and its putative role in regulating fatty acid oxidation. Am J Physiol 1998; 275: H2122–H2129.
Kudo N, Barr AJ, Barr RL, Desai S, Lopaschuk GD . High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5'-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J Biol Chem 1995; 270: 17513–17520.
Koutsari C, Jensen MD . Thematic review series: patient-oriented research. Free fatty acid metabolism in human obesity. J Lipid Res 2006; 47: 1643–1650.
Peterson LR, Herrero P, Schechtman KB, Racette SB, Waggoner AD, Kisrieva-Ware Z et al. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation 2004; 109: 2191–2196.
Lopaschuk GD, Russell JC . Myocardial function and energy substrate metabolism in the insulin-resistant JCR: LA corpulent rat. J Appl Physiol 1991; 71: 1302–1308.
Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED . Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 2005; 112: 2686–2695.
Gibbs JW . A method of geometrical representation of the thermodynamic properties of substances by means of surfaces. Transactions of the Connecticut Academy vol 2. Connecticut Academy of Arts and Sciences: New Haven, 1873. pp 382–404.
Alberty RA . Standard Gibbs free energy, enthalpy, and entropy changes as a function of Ph and Pmg for several reactions involving adenosine phosphates. J Biol Chem 1969; 244: 3290–3302.
Veech RL . The determination of the redox states and phosphorylation potential in living tissues and their relationship to metabolic control of disease phenotypes. Biochem Mol Biol Edu 2006; 34: 168–179.
Stubbs M, Veech R, Krebs H . Control of the redox state of the nicotinamide–adenine dinucleotide couple in rat liver cytoplasm. Biochem J 1972; 126: 59.
Krebs H, Veech R . Equilibrium relations between pyridine nucleotides and adenine nucleotides and their roles in the regulation of metabolic processes. Adv Enzyme Regul 1969; 7: 397–413.
Veech R, Raijman L, Krebs H . Equilibrium relations between the cytoplasmic adenine nucleotide system and nicotinamide–adenine nucleotide system in rat liver. Biochem J 1970; 117: 499.
Veech RL, Eggleston LV, Krebs HA . The redox state of free nicotinamide-adenine dinucleotide phosphate in the cytoplasm of rat liver. Biochem J 1969; 115: 609–619.
Veech RL, Kashiwaya Y, Gates DN, King MT, Clarke K . The energetics of ion distribution: the origin of the resting electric potential of cells. IUBMB Life 2002; 54: 241–252.
Kashiwaya Y, King MT, Veech RL . Substrate signaling by insulin: a ketone bodies ratio mimics insulin action in heart. Am J Cardiol 1997; 80: 50A–64A.
Wilson DF, Stubbs M, Veech RL, Erecińska M, Krebs HA . Equilibrium relations between the oxidation–reduction reactions and the adenosine triphosphate synthesis in suspensions of isolated liver cells. Biochem J 1974; 140: 57.
Veech RL, Lawson J, Cornell N, Krebs HA . Cytosolic phosphorylation potential. J Biol Chem 1979; 254: 6538.
Veech RL, Fell DA . Distribution control of metabolic flux. Cell Biochem Funct 1996; 14: 229–236.
Veech RL . The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leuko Essent Fatty Acids 2004; 70: 309–319.
Mitchell PD . Chemiosmotic Coupling and Energy Transduction. Glynn Research: Bodmin, Cornwall, 1968.
Veech RL, Chance B, Kashiwaya Y, Lardy HA, Cahill GR . Ketone bodies, potential therapeutic uses. IUBMB Life 2001; 51: 241–247.
Hue L, Taegtmeyer H . The Randle cycle revisited: a new head for an old hat. Am J Physiol-Endoc Metab 2009; 297: E578–E591.
Yasuda R, Noji H, Kinosita K, Yoshida M . F-1-ATPase is a highly efficient molecular motor that rotates with discrete 120 degrees steps. Cell 1998; 93: 1117–1124.
Veech RL . The determination of the redox states and phosphorylation potential in living tissues and their relationship to metabolic control of disease phenotypes. Biochem Mol Biol Educ 2006; 34: 168–179.
Cole MA, Murray AJ, Cochlin LE, Heather LC, McAleese S, Knight NS et al. A high fat diet increases mitochondrial fatty acid oxidation and uncoupling to decrease efficiency in rat heart. Basic Res Cardiol 2011; 106: 447–457.
Korvald C, Elvenes OP, Myrmel T . Myocardial substrate metabolism influences left ventricular energetics in vivo. Am J Physiol Heart Circ Physiol 2000; 278: H1345–H1351.
Lopaschuk GD, Ussher JR, Folmes CDL, Jaswal JS, Stanley WC . Myocardial fatty acid metabolism in health and disease. Physiol Rev 2010; 90: 207–258.
Neubauer S . Mechanisms of disease - the failing heart - an engine out of fuel. New Engl J Med 2007; 356: 1140–1151.
Peterson LR, Soto PF, Herrero P, Mohammed BS, Avidan MS, Schechtman KB et al. Impact of gender on the myocardial metabolic response to obesity. JACC Cardiovasc Imaging 2008; 1: 424–433.
Wilson CR, Tran MK, Salazar KL, Young ME, Taegtmeyer H . Western diet, but not high fat diet, causes derangements of fatty acid metabolism and contractile dysfunction in the heart of Wistar rats. Biochem J 2007; 406: 457–467.
Murray AJ, Anderson RE, Watson GC, Radda GK, Clarke K . Uncoupling proteins in human heart. Lancet 2004; 364: 1786–1788.
Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 2007; 56: 2457–2466.
Kolter T, Uphues I, Eckel J . Molecular analysis of insulin resistance in isolated ventricular cardiomyocytes of obese Zucker rats. Am J Physiol 1997; 273: E59–E67.
Randle PJ, Kerbey AL, Espinal J . Mechanisms decreasing glucose oxidation in diabetes and starvation: role of lipid fuels and hormones. Diabetes Metab Rev 1988; 4: 623–638.
Tsutsumi E, Takenaka F . Inhibition of pyruvate kinase by free fatty acids in rat heart muscle. Biochim Biophys Acta 1969; 171: 355–357.
Priestman DA, Orfali KA, Sugden MC . Pyruvate inhibition of pyruvate dehydrogenase kinase. Effects of progressive starvation and hyperthyroidism in vivo, and of dibutyryl cyclic AMP and fatty acids in cultured cardiac myocytes. FEBS Lett 1996; 393: 174–178.
Bryson JM, Cooney GJ, Wensley VR, Phuyal JL, Caterson ID . The effects of the inhibition of fatty acid oxidation on pyruvate dehydrogenase complex activity in tissues of lean and obese mice. Int J Obes Relat Metab Disord 1996; 20: 738–744.
Schroeder MA, Atherton HJ, Heather LC, Griffin JL, Clarke K, Radda GK et al. Determining the in vivo regulation of cardiac pyruvate dehydrogenase based on label flux from hyperpolarised [1-13C]pyruvate. NMR Biomed 2011; 24: 980–987.
Schroeder MA, Cochlin LE, Heather LC, Clarke K, Radda GK, Tyler DJ . In vivo assessment of pyruvate dehydrogenase flux in the heart using hyperpolarized carbon-13 magnetic resonance. Proc Natl Acad Sci USA 2008; 105: 12051–12056.
Atherton HJ, Schroeder MA, Dodd MS, Heather LC, Carter EE, Cochlin LE et al. Validation of the in vivo assessment of pyruvate dehydrogenase activity using hyperpolarised 13C MRS. NMR Biomed 2011; 24: 201–208.
Li J, Feuers RJ, Desai VG, Lewis SM, Duffy PH, Mayhugh MA et al. Surgical caloric restriction ameliorates mitochondrial electron transport dysfunction in obese females. Obes Surg 2007; 17: 800–808.
Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE . Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 2005; 54: 8–14.
Shelley P, Martin-Gronert MS, Rowlerson A, Poston L, Heales SJ, Hargreaves IP et al. Altered skeletal muscle insulin signaling and mitochondrial complex II-III linked activity in adult offspring of obese mice. Am J Physiol Regul Integr Comp Physiol 2009; 297: R675–R681.
Chanseaume E, Barquissau V, Salles J, Aucouturier J, Patrac V, Giraudet C et al. Muscle mitochondrial oxidative phosphorylation activity, but not content, is altered with abdominal obesity in sedentary men: synergism with changes in insulin sensitivity. J Clin Endocrinol Metab 2010; 95: 2948–2956.
Lee L, Campbell R, Scheuermann-Freestone M, Taylor R, Gunaruwan P, Williams L et al. Metabolic modulation with perhexiline in chronic heart failure: a randomized, controlled trial of short-term use of a novel treatment. Circulation 2005; 112: 3280–3288.
Wu L, Belardinelli L, Fraser H . A novel partial fatty acid oxidation inhibitor decreases myocardial oxygen consumption and improves cardiac efficiency in demand-induced ischemic heart. J Cardiovasc Pharmacol 2008; 51: 372–379.
Mouquet F, Rousseau D, Domergue-Dupont V, Grynberg A, Liao R . Effects of trimetazidine, a partial inhibitor of fatty acid oxidation, on ventricular function and survival after myocardial infarction and reperfusion in the rat. Fundam Clin Pharmacol 2010; 24: 469–476.
Maarman G, Marais E, Lochner A, du Toit EF . Effect of chronic CPT-1 inhibition on myocardial ischemia-reperfusion injury (I/R) in a model of diet-induced obesity. Cardiovasc Drugs Ther 2012; 26: 205–216.
Nakae I, Mitsunami K, Omura T, Yabe T, Tsutamoto T, Matsuo S et al. Proton magnetic resonance spectroscopy can detect creatine depletion associated with the progression of heart failure in cardiomyopathy. J Am Coll Cardiol 2003; 42: 1587–1593.
Ingwall JS, Atkinson DE, Clarke K, Fetters JK . Energetic correlates of cardiac-failure - changes in the creatine-kinase system in the failing myocardium. Eur Heart J 1990; 11: 108–115.
Rider O, Francis J, Ali M, Holloway C, Pegg T, Robson M et al. Effects of catecholamine stress on diastolic function and myocardial energetics in obesity. Circulation 2012; 125: 1511–1519.
Perseghin G, Ntali G, De Cobelli F, Lattuada G, Esposito A, Belloni E et al. Abnormal left ventricular energy metabolism in obese men with preserved systolic and diastolic functions is associated with insulin resistance. Diabetes Care 2007; 30: 1520–1526.
Stankov MV, Lucke T, Das AM, Schmidt RE, Behrens GM . Relationship of mitochondrial DNA depletion and respiratory chain activity in preadipocytes treated with nucleoside reverse transcriptase inhibitors. Antivir Ther 2007; 12: 205–216.
Berger J, Moller DE . The mechanisms of action of PPARs. Annu Rev Med 2002; 53: 409–435.
Huss JM, Kelly DP . Nuclear receptor signaling and cardiac energetics. Circ Res 2004; 95: 568–578.
Chandran M, Phillips SA, Ciaraldi T, Henry RR . Adiponectin: more than just another fat cell hormone? Diabetes Care 2003; 26: 2442–2450.
Barger PM, Kelly DP . PPAR signaling in the control of cardiac energy metabolism. Trends Cardiovasc Med 2000; 10: 238–245.
Motojima K, Passilly P, Peters JM, Gonzalez FJ, Latruffe N . Expression of putative fatty acid transporter genes are regulated by peroxisome proliferator-activated receptor alpha and gamma activators in a tissue- and inducer-specific manner. J Biol Chem 1998; 273: 16710–16714.
Van Bilsen M, de Vries JE, Van der Vusse GJ . Long-term effects of fatty acids on cell viability and gene expression of neonatal cardiac myocytes. Prostaglandins Leukot Essent Fatty Acids 1997; 57: 39–45.
van der Lee KA, Vork MM, De Vries JE, Willemsen PH, Glatz JF, Reneman RS et al. Long-chain fatty acid-induced changes in gene expression in neonatal cardiac myocytes. J Lipid Res 2000; 41: 41–47.
Djouadi F, Brandt JM, Weinheimer CJ, Leone TC, Gonzalez FJ, Kelly DP . The role of the peroxisome proliferator-activated receptor alpha (PPAR alpha) in the control of cardiac lipid metabolism. Prostaglandins Leukot Essent Fatty Acids 1999; 60: 339–343.
Pagano C, Calcagno A, Granzotto M, Calabrese F, Thiene G, Federspil G et al. Heart lipid accumulation in obese non-diabetic rats: effect of weight loss. Nutr Metab Cardiovasc Dis 2008; 18: 189–197.
Bartels ED, Nielsen JM, Hellgren LI, Ploug T, Nielsen LB . Cardiac expression of microsomal triglyceride transfer protein is increased in obesity and serves to attenuate cardiac triglyceride accumulation. PloS One 2009; 4: e5300.
Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P et al. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest 2001; 107: 813–822.
Young ME, McNulty P, Taegtmeyer H . Adaptation and maladaptation of the heart in diabetes: Part II: potential mechanisms. Circulation 2002; 105: 1861–1870.
Taegtmeyer H, McNulty P, Young ME . Adaptation and maladaptation of the heart in diabetes: Part I: general concepts. Circulation 2002; 105: 1727–1733.
Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A et al. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci USA 2003; 100: 1226–1231.
Distel RJ, Robinson GS, Spiegelman BM . Fatty acid regulation of gene expression. Transcriptional and post-transcriptional mechanisms. J Biol Chem 1992; 267: 5937–5941.
van der Vusse GJ, Glatz JF, Stam HC, Reneman RS . Fatty acid homeostasis in the normoxic and ischemic heart. Physiol Rev 1992; 72: 881–940.
Luiken JJ, Arumugam Y, Dyck DJ, Bell RC, Pelsers MM, Turcotte LP et al. Increased rates of fatty acid uptake and plasmalemmal fatty acid transporters in obese Zucker rats. J Biol Chem 2001; 276: 40567–40573.
Coort SL, Hasselbaink DM, Koonen DP, Willems J, Coumans WA, Chabowski A et al. Enhanced sarcolemmal FAT/CD36 content and triacylglycerol storage in cardiac myocytes from obese zucker rats. Diabetes 2004; 53: 1655–1663.
Brindley DN, Kok BP, Kienesberger PC, Lehner R, Dyck JR . Shedding light on the enigma of myocardial lipotoxicity: the involvement of known and putative regulators of fatty acid storage and mobilization. Am J Physiol 298: E897–E908.
Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D et al. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci USA 2000; 97: 1784–1789.
Ahima RS . Adipose tissue as an endocrine organ. Obesity (Silver Spring, Md) 2006; 14: 242S–249S.
Li L, Wu L, Wang C, Liu L, Zhao Y . Adiponectin modulates carnitine palmitoyltransferase-1 through AMPK signaling cascade in rat cardiomyocytes. Regul Pept 2007; 139: 72–79.
Palanivel R, Fang X, Park M, Eguchi M, Pallan S, De Girolamo S et al. Globular and full-length forms of adiponectin mediate specific changes in glucose and fatty acid uptake and metabolism in cardiomyocytes. Cardiovasc Res 2007; 75: 148–157.
Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 1999; 257: 79–83.
Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med 1996; 334: 292–295.
Lopaschuk GD, Folmes CD, Stanley WC . Cardiac energy metabolism in obesity. Circ Res 2007; 101: 335–347.
Unger RH, Zhou YT, Orci L . Regulation of fatty acid homeostasis in cells: novel role of leptin. Proc Natl Acad Sci USA 1999; 96: 2327–2332.
Viljanen AP, Karmi A, Borra R, Parkka JP, Lepomaki V, Parkkola R et al. Effect of caloric restriction on myocardial fatty acid uptake, left ventricular mass, and cardiac work in obese adults. Am J Cardiol 2009; 103: 1721–1726.
Lin CH, Kurup S, Herrero P, Schechtman KB, Eagon JC, Klein S et al. Myocardial oxygen consumption change predicts left ventricular relaxation improvement in obese humans after weight loss. Obesity (Silver Spring) 2011; 19: 1804–1812.
Utz W, Engeli S, Haufe S, Kast P, Bohnke J, Haas V et al. Moderate dietary weight loss reduces myocardial steatosis in obese and overweight women. Int J Cardiol 2012; e-pub ahead of print 9 April 2012.
Rider O, Pegg TJ, Robson MD, Tyler DJ, Byrne JP, Clarke K et al. The effects of catecholamine stress and weight loss on myocardial relaxation and high energy phosphate metabolism in obesity. Circulation 2009; 120: s405 Abstract 839.
Algahim MF, Sen S, Taegtmeyer H . Bariatric surgery to unload the stressed heart: a metabolic hypothesis. Am J Physiol Heart Circ Physiol 2012; 302: H1539–H1545.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Rider, O., Cox, P., Tyler, D. et al. Myocardial substrate metabolism in obesity. Int J Obes 37, 972–979 (2013). https://doi.org/10.1038/ijo.2012.170
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijo.2012.170
Keywords
This article is cited by
-
Temporal relationships between BMI and obesity-related predictors of cardiometabolic and breast cancer risk in a longitudinal cohort
Scientific Reports (2023)
-
Evaluation of left ventricular function in obese patients with obstructive sleep apnea by three-dimensional speckle tracking echocardiography
The International Journal of Cardiovascular Imaging (2022)
-
Strategies for Imaging Metabolic Remodeling of the Heart in Obesity and Heart Failure
Current Cardiology Reports (2022)
-
Epicardial Adipose Tissue and Cardiovascular Disease
Current Hypertension Reports (2019)
-
Diabetic Cardiomyopathy Modelling Using Induced Pluripotent Stem Cell Derived Cardiomyocytes: Recent Advances and Emerging Models
Stem Cell Reviews and Reports (2019)
