
Abstract
The replication of genome-wide significant association signals in independent populations is a practical approach for characterizing gene-disease relationships. Therefore, we sought to explore the top 10 polymorphisms from a large blood pressure genome-wide association study of northeastern Han Chinese East Asians. This was a hospital-based study involving 1009 patients with essential hypertension and 756 normotensive controls from Qiqihar city, China. Genotyping was conducted with a polymerase chain reaction-ligase detection reaction method. All polymorphisms except for rs6825911 satisfied Hardy–Weinberg equilibrium. Overall, the genotype differences between the patients and controls were significant for rs35444 (P<0.001), rs11191548 (P=0.017) and rs17249754 (P=0.017). The per-minor-allele odds ratios of rs35444, rs11191548 and rs17249754 were 0.54 (95% confidence interval (95% CI): 0.46–0.62; P<0.01), 1.23 (95% CI: 1.07–1.43; P=0.005) and 1.23 (95% CI: 1.07–1.41; P=0.004), respectively. Similarly, the carriers of minor homozygotes had a significant reduction in adjusted systolic and diastolic blood pressure for rs35444 (P<0.01) but an increase for both rs11191548 (P<0.01) and rs17249754 (P<0.04). Further application of the genetic risk score method indicated that subjects with risk scores of 8, 10 and 12–16 had 1.66-fold (95% CI: 1.01–2.72), 1.72-fold (95% CI: 1.03–2.86) and 1.97-fold (95% CI: 1.12–3.46) increases, respectively, in the odds of developing hypertension, and similar increases were also observed for blood pressure. Taken together, our findings demonstrate that although only three of the top 10 polymorphisms were successfully validated in the northeastern Han Chinese population, the genetic risk score analyses led us to more profound insights into the possible joint effects of multiple polymorphisms on hypertension risk and blood pressure variation.
Similar content being viewed by others


Introduction
Despite the unbiased and hypothesis-free merits of genome-wide association (GWA) studies, the challenge of understanding the ultimate genetic architecture of common polygenic diseases and quantitative phenotypes has eluded global geneticists for decades.1 The most likely reason for this is genetic heterogeneity across ethnic groups.2 The replication of genome-wide significant association signals in independent populations is a practical approach for characterizing gene-disease relationships.3 We have recently explored the contributory roles of 13 top hypertension-susceptibility polymorphisms from two GWA studies of populations of non-Asian descent4, 5 and found that these polymorphisms collectively explained only a small fraction of the heritable components in Chinese subjects. In addition to the potential existence of genetic heterogeneity between Asian and non-Asian populations, other possibilities such as the disregarding of combined genetic effects and lack of consideration of confounders, might explain, at least in part, the divergence of these findings.6 To elucidate this issue, in this study, we sought to explore the top 10 polymorphisms from a large blood pressure genome-wide association study of East Asians,7 in northeastern Han Chinese population.
Methods
Study population
This was a hospital-based case–control study incorporating 1765 unrelated participants from Qiqihar city, Heilongjiang province, China. All participants were of Han Chinese descent and local residents of Qiqihar city in northeast China. In addition, the participants were classified into an essential hypertension group and normotensive control group according to clinical and laboratory examinations. The institutional review board of Qiqihar Medical University approved this study, and each participant provided informed written consent at enrollment. This study was conducted according to the Declaration of Helsinki Principles.
Essential hypertension was defined as a mean systolic blood pressure more than 140 mm Hg or a diastolic blood pressure more than 90 mm Hg or the current use of antihypertensive drugs. Blood pressure was measured using a calibrated mercury sphygmomanometer with an appropriate adult cuff size by certified examiners. Patients with clinical evidence of secondary hypertension and renal disease were excluded. The essential hypertension group contained 1009 sporadic patients aged 64.48 (s.d.: 8.53) years, and 54.31% of the patients were men. The remaining participants (n=756), who had normal blood pressure, formed the age- and gender-matched control group.
Demographic and clinical measurement
Demographic data including age, gender, body weight and height were recorded at enrollment. Body weight was recorded in light clothing, and body mass index (BMI) was calculated as weight in kilograms divided by height in meters squared. Blood samples were collected after overnight fasting, and patient sera were isolated immediately. The plasma levels of triglyceride, total cholesterol, high-density lipoprotein cholesterol, low-density lipoprotein cholesterol, blood urea nitrogen, creatinine and urea acid were determined enzymatically using available kits and an auto analyzer.
Genotyping
Genomic DNA was isolated from peripheral blood leukocytes according to a standard phenol-chloroform method and was stored at −40 °C until required for batch genotyping. The genotypes of the 10 examined polymorphisms, including rs17030613, rs16849225, rs6825911, rs1173766, rs11066280, rs35444, rs880315, rs16998073, rs11191548 and rs17249754, were determined by a polymerase chain reaction-ligase detection reaction (PCR-LDR) method as previously described.8 The primers for amplification and the probes for the LDR are presented in Supplementary Table 1.
The PCR primers were synthesized by Shanghai Generay Biotech Co., Ltd., and the PCR reactions were conducted in an EDC-810 Amplifier (Dongsheng Innovation Biotech Co., Ltd., Beijing, China). The cycling parameters were as follows: 94 °C for 2 min; 35 cycles of 94 °C for 20 s, 60 °C for 20 s, and 72 °C for 20 s; and a final extension step at 72 °C for 3 min. For each polymorphism, two specific probes were synthesized to discriminate specific bases with one common probe labeled with 6-carboxy-fluorescein at the 3′-end and phosphorylated at the 5′- end. The multiplex ligation reaction was conducted in a reaction volume of 10 μl containing 2 μl of PCR product, 1 μl of 10 × Taq DNA ligase buffer, 1 μM of each discriminating probe and 5 U of Taq DNA ligase. The ligation parameters were 30 cycles of 94 °C for 30 s and 56 °C for 3 min. After the reaction, 1 μl of LDR reaction product was mixed with 1 μl of ROX passive reference and 1 μl of loading buffer before being denatured at 95 °C for 3 min and chilled rapidly in ice water. The fluorescent products of the LDR were differentiated using an ABI 3730XL sequencer (Applied Biosystems, CA, USA). To test the accuracy of the PCR-LDR method, we randomly selected 96 samples as an internal reference in batch genotyping, and the concordance of these samples reached 100%.
Statistical analysis
Statistical analyses were conducted with the STATA software version 11.2 (StataCorp LP, College Station, TX, USA) in Windows. The study power was calculated using the PS (Power and Sample Size Calculations) software version 3.0. A two-tailed P<0.05 was considered to indicate statistical significance.
Quantitative variables were compared between the patients and controls with Student’s t-test. The goodness-of-fit of the observed allele frequencies with the expected frequencies according to the Hardy–Weinberg equilibrium and genotype/allele frequencies between the two groups were computed with the χ2-test. The per-minor-allele odds ratio (OR) of each polymorphism was evaluated using the genetic risk score method,9, 10 which assigns scores of 0, 1 and 2 to major homozygotes, heterozygotes and minor homozygotes, respectively, and by calculating the OR per unit score by logistic regression before and after adjusting for age, gender and BMI.
As recommended by Tobin et al.,11 for participants under antihypertensive medication, blood pressure was determined by adding 15 mm Hg and 10 mm Hg to the systolic and diastolic blood pressure, respectively, and this method was in agreement with the study by Newton-Cheh et al.12 Unless otherwise indicated, the adjusted systolic and diastolic blood pressures were analyzed.
Assuming that an individual polymorphism with a modest effect on blood pressure has insufficient power to exert an effect on incident hypertension events, a genotype risk score was therefore created on the basis of the total number of risk-conferring alleles of each polymorphism in Hardy–Weinberg equilibrium by assigning a score 0, 1 and 2 to the risk-reducing genotype, heterozygous genotype and risk-conferring genotype, respectively. For example, if we assess nine bi-allelic polymorphisms, the genetic risk score will theoretically range from 0 to 18.
Results
Baseline characteristics
Demographic and clinical data are compared between the hypertensive patients and normotensive controls in Table 1. The distributions of age (P=0.751) and gender (P=0.843) were comparable between the two groups. However, BMI (P<0.001), blood pressure (both P<0.001), fasting glucose (P<0.001), triglycerides (P=0.024) and creatinine (P=0.002) levels were significantly higher in patients than in controls (P<0.05), but the high-density lipoprotein cholesterol values were lower in patients than controls (P<0.001).
Single-polymorphism analysis
Hardy–Weinberg equilibrium was satisfied in controls for all 10 polymorphisms except for rs6825911 (P<0.01). The genotype distributions and allele frequencies of the 10 polymorphisms are summarized in Table 2. Overall, there were significant genotype differences for rs35444 (P<0.001), rs11191548 (P=0.017) and rs17249754 (P=0.017) between patients and controls. Under the additive model, the per-minor-allele ORs of rs35444, rs11191548 and rs17249754 were 0.54 (95% CI: 0.46–0.62; P<0.01), 1.23 (95% CI: 1.07–1.43; P=0.005) and 1.23 (95% CI: 1.07–1.41; P=0.004), respectively, after adjusting for confounding factors. Correspondingly, the sample size in this study provided 100%, 83.3% and 82.2% power to detect significant ORs of 0.54, 1.23 and 1.23 between the two groups, respectively.
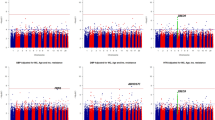
The distributions of the adjusted blood pressures across the genotypes of three significant polymorphisms in hypertensive patients are presented in Figure 1. After assigning the major homozygotes of each polymorphism as the reference group, the carriers of minor homozygotes had a significant reduction in adjusted systolic and diastolic blood pressures for rs35444 (both P<0.01) but an increase for rs11191548 (both P<0.01) and rs17249754 (both P<0.04).

The distributions of adjusted systolic and diastolic blood pressure across the genotypes of three significant polymorphisms in hypertensive patients.
Genetic risk score analysis
Because of the distortion of Hardy–Weinberg equilibrium for rs6825911, the genetic risk score was created for the other nine polymorphisms based on the total number of risk-conferring alleles in all participants with total scores ranging from 2 to 16 (Table 3). The risk-conferring allele was defined according to the ORs in Table 2, that is, the risk-conferring allele refers to the minor allele if the per-allele OR was greater than the unity; otherwise, the risk-conferring allele was the major allele. Because of the low penetrance, participants carrying 2–5 and 12–16 risk-conferring alleles were combined as single groups.
Overall, there was a roughly graded increase in the risk of developing hypertension with increasing genetic risk scores. After assigning those with a score range 2–5 as the reference group, we found that subjects with scores of 8, 10 and 12–16 had significant 1.66-fold (95% CI: 1.01–2.72; P=0.045), 1.72-fold (95% CI: 1.03–2.86; P=0.036) and 1.97-fold (95% CI: 1.12–3.46; 0.019) increases in risk for hypertension, and the significance was preserved after adjusting for confounders. Similarly, with the increase of genetic risk scores, the levels of adjusted blood pressure were gradually increased, especially for carriers of eight or more risk-conferring alleles in hypertensive patients. For example, compared with the score range 2–5, the carriers with 8, 10 and 12–16 scores had 6.73, 7.71 and 14.47 mm Hg increases in adjusted systolic blood pressure, respectively, and 4.8, 6.92 and 7.09 increases in adjusted diastolic blood pressure, respectively (P<0.01 for all).
Discussion
The aim of this study was to replicate, in northeastern Han Chinese, the top 10 polymorphisms of a large blood pressure GWA study7 involving 50 373 participants of East Asian descent. It is worth noting that three of the top 10 polymorphisms were observed to exhibit a high degree of association with both hypertension and blood pressure in this study. However, we cannot rule out the contributory role of the other seven polymorphisms in the pathogenesis of hypertension because our genetic risk score analyses led us to more profound insights into the possible joint role of multiple polymorphisms in entailing hypertension risk and blood pressure variation.
Waves of genetic association studies are coming at us like a tsunami, with a deluge of data regarding the identification and characterization of genes or variants contributing to common complex diseases.13, 14 Geneticists urgently need to tease out the causative genetic effects.15, 16 Although only a small fraction of the top polymorphisms with genomic significance have been successfully replicated in this study, our findings offer some potential hints. Hypertension is a multifactorial disorder in which genetic factors account for 30 to 50% of blood pressure variation.17, 18 It is generally believed that the relative risk attributable to a single locus is small. As exemplified in the GWA study of East Asians,7 the per-allele increase of rs35444 resulted in an increase of 0.63 and 0.5 mm Hg in systolic and diastolic blood pressures, respectively.7 Contrastingly, in the present study, we observed that relative to the rs35444-TT genotype carriers, the adjusted systolic and diastolic blood pressure of those with the rs35444-CC genotype increased by 10.8 and 5.7 mm Hg, respectively. Although residual confounding by incompletely measured or unmeasured physiologic covariates might exist in our findings, it appears unlikely that our results could be explained by confounders.
Genetic heterogeneity might also be a possible explanation for the discrepancy of these observations.6 China has a vast territory, and geographic differences cause various discrepancies as a result of historical and cultural impact. For example, salt consumption between northern and southern Chinese differs remarkably. Indeed, on the grounds of a multi-center observation in China, the average urinary sodium excretion in northern Chinese (271 mmol per day) is nearly double the amount of that in southern Chinese (139 mmol per day), leading to an average of 7.4 and 6.9 mm Hg increases in systolic and diastolic blood pressure, respectively.19 As reflected in this study, the minor allele frequencies of rs16998073 were 40.34% in hypertensive patients and 39.81% in normotensive controls, much higher than that observed in the GWA study of East Asians as a whole (30%). Genetic heterogeneity is an inevitable problem in disease identification strategies and can be somewhat avoided by analyzing homogeneous populations. In fact, our study population is characterized by genetic homogeneity and geographic stability, and the subjects are most likely uniform in their environmental exposures, including habitual dietary intake of high salt and high fat20 and a lower rate of hypertension recognition and treatment, rendering this population more appropriate for enhancing our understanding of the genetic predisposition to essential hypertension.
Despite the fact that seven of the examined polymorphisms failed to reach statistical significance, we cannot rule out the contributory roles of these polymorphisms in the pathogenesis of hypertension because the further application of the genetic risk score method detected an approximate linear trend for both hypertension as a binary trait and blood pressure as a quantitative trait, which supports the claim that the increasing risk conferred by a single locus is small. Genetic risk score, a type of summary of the cumulative effect of individual polymorphisms, can enhance the detection of persons at risk for hypertension.21 However, because of the assumption that each allele in the risk score calculation is treated as equal, our findings must be considered preliminary and should be viewed as hypothesis-generating for future large and well-designed studies.
Interpretation of our results, however, should be viewed in light of several limitations. First, this study was retrospective in design, which precludes further comments on the cause-effect relationship.22 Second, we only focused on the top 10 polymorphisms from a large blood pressure GWA study of East Asians, and this resulted in the incorporation of more functional polymorphisms, especially the low-penetrance polymorphisms from promising hypertension-susceptibility genes, such as the apelin-AGTRL1 pathway genes.15, 23 More importantly, because hypertension is a multifactorial disease,24 characterizing the interaction of different polymorphisms from different chromosomes is regarded as an effective approach to elucidate the pathogenesis of hypertension. Third, the fact that our study participants were of northeastern Han Chinese descent limited the generalizability of our findings, necessitating further confirmation in other ethnic populations.
Taken together, our findings demonstrate that, although only a small fraction of the top polymorphisms with genomic significance were successfully validated in this study, our genetic risk score analyses led us to more profound insights into the possible joint effects of multiple polymorphisms on hypertension risk and blood pressure variation. Nevertheless, for practical reasons, we hope that this study will not remain just another end point of research but instead would encourage more validation studies of promising GWA results in other independent, genetically homogeneous populations, which would aid in acquiring a better understanding of the genetic underpinnings of essential hypertension.
References
Gayan J, Gonzalez-Perez A, Bermudo F, Saez ME, Royo JL, Quintas A, Galan JJ, Moron FJ, Ramirez-Lorca R, Real LM, Ruiz A . A method for detecting epistasis in genome-wide studies using case-control multi-locus association analysis. BMC Genomics 2008; 9: 360.
Wang X, Chua HX, Chen P, Ong RT, Sim X, Zhang W, Takeuchi F, Liu X, Khor CC, Tay WT, Cheng CY, Suo C, Ng DP, Liu J, Aung T, Chia KS, Kooner JS, Chambers JC, Wong TY, Tai ES, Kato N, Teo YY . Comparing methods for performing trans-ethnic meta-analysis of genome-wide association studies. Hum Mol Genet 2013; 22: 2303–2311.
Chanock SJ, Manolio T, Boehnke M, Boerwinkle E, Hunter DJ, Thomas G, Hirschhorn JN, Abecasis G, Altshuler D, Bailey-Wilson JE, Brooks LD, Cardon LR, Daly M, Donnelly P, Fraumeni JF Jr., Freimer NB, Gerhard DS, Gunter C, Guttmacher AE, Guyer MS, Harris EL, Hoh J, Hoover R, Kong CA, Merikangas KR, Morton CC, Palmer LJ, Phimister EG, Rice JP, Roberts J, Rotimi C, Tucker MA, Vogan KJ, Wacholder S, Wijsman EM, Winn DM, Collins FS . Replicating genotype-phenotype associations. Nature 2007; 447: 655–660.
Niu W, Zhang Y, Ji K, Gu M, Gao P, Zhu D . Confirmation of top polymorphisms in hypertension genome wide association study among Han Chinese. Clin Chim Acta 2010; 411: 1491–1495.
Niu WQ, Zhang Y, Ji KD, Gao PJ, Zhu DL . Contribution of five top whole-genome association signals to hypertension in Han Chinese. J Hum Hypertens 2011; 25: 278–280.
Kato N . Ethnic differences in genetic predisposition to hypertension. Hypertens Res 2012; 35: 574–581.
Kato N, Takeuchi F, Tabara Y, Kelly TN, Go MJ, Sim X, Tay WT, Chen CH, Zhang Y, Yamamoto K, Katsuya T, Yokota M, Kim YJ, Ong RT, Nabika T, Gu D, Chang LC, Kokubo Y, Huang W, Ohnaka K, Yamori Y, Nakashima E, Jaquish CE, Lee JY, Seielstad M, Isono M, Hixson JE, Chen YT, Miki T, Zhou X, Sugiyama T, Jeon JP, Liu JJ, Takayanagi R, Kim SS, Aung T, Sung YJ, Zhang X, Wong TY, Han BG, Kobayashi S, Ogihara T, Zhu D, Iwai N, Wu JY, Teo YY, Tai ES, Cho YS, He J . Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nat Genet 2011; 43: 531–538.
Khanna M, Park P, Zirvi M, Cao W, Picon A, Day J, Paty P, Barany F . Multiplex PCR/LDR for detection of K-ras mutations in primary colon tumors. Oncogene 1999; 18: 27–38.
Thanassoulis G, Peloso GM, Pencina MJ, Hoffmann U, Fox CS, Cupples LA, Levy D, D'Agostino RB, Hwang SJ, O'Donnell CJ . A genetic risk score is associated with incident cardiovascular disease and coronary artery calcium: the Framingham Heart Study. Circ Cardiovasc Genet 2012; 5: 113–121.
Chen H, Poon A, Yeung C, Helms C, Pons J, Bowcock AM, Kwok PY, Liao W . A genetic risk score combining ten psoriasis risk loci improves disease prediction. PLoS One 2011; 6: e19454.
Tobin MD, Sheehan NA, Scurrah KJ, Burton PR . Adjusting for treatment effects in studies of quantitative traits: antihypertensive therapy and systolic blood pressure. Stat Med 2005; 24: 2911–2935.
Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, Najjar SS, Zhao JH, Heath SC, Eyheramendy S, Papadakis K, Voight BF, Scott LJ, Zhang F, Farrall M, Tanaka T, Wallace C, Chambers JC, Khaw KT, Nilsson P, van der Harst P, Polidoro S, Grobbee DE, Onland-Moret NC, Bots ML, Wain LV, Elliott KS, Teumer A, Luan J, Lucas G, Kuusisto J, Burton PR, Hadley D, McArdle WL, Brown M, Dominiczak A, Newhouse SJ, Samani NJ, Webster J, Zeggini E, Beckmann JS, Bergmann S, Lim N, Song K, Vollenweider P, Waeber G, Waterworth DM, Yuan X, Groop L, Orho-Melander M, Allione A, Di Gregorio A, Guarrera S, Panico S, Ricceri F, Romanazzi V, Sacerdote C, Vineis P, Barroso I, Sandhu MS, Luben RN, Crawford GJ, Jousilahti P, Perola M, Boehnke M, Bonnycastle LL, Collins FS, Jackson AU, Mohlke KL, Stringham HM, Valle TT, Willer CJ, Bergman RN, Morken MA, Doring A, Gieger C, Illig T, Meitinger T, Org E, Pfeufer A, Wichmann HE, Kathiresan S, Marrugat J, O'Donnell CJ, Schwartz SM, Siscovick DS, Subirana I, Freimer NB, Hartikainen AL, McCarthy MI, O'Reilly PF, Peltonen L, Pouta A, de Jong PE, Snieder H, van Gilst WH, Clarke R, Goel A, Hamsten A, Peden JF, Seedorf U, Syvanen AC, Tognoni G, Lakatta EG, Sanna S, Scheet P, Schlessinger D, Scuteri A, Dorr M, Ernst F, Felix SB, Homuth G, Lorbeer R, Reffelmann T, Rettig R, Volker U, Galan P, Gut IG, Hercberg S, Lathrop GM, Zelenika D, Deloukas P, Soranzo N, Williams FM, Zhai G, Salomaa V, Laakso M, Elosua R, Forouhi NG, Volzke H, Uiterwaal CS, van der Schouw YT, Numans ME, Matullo G, Navis G, Berglund G, Bingham SA, Kooner JS, Connell JM, Bandinelli S, Ferrucci L, Watkins H, Spector TD, Tuomilehto J, Altshuler D, Strachan DP, Laan M, Meneton P, Wareham NJ, Uda M, Jarvelin MR, Mooser V, Melander O, Loos RJ, Elliott P, Abecasis GR, Caulfield M, Munroe PB . Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet 2009; 41: 666–676.
Katsuya T, Harrap SB, Ogihara T . Genetics of hypertension and cardiovascular disease. Int J Hypertens 2011; 2010: 951254.
Weiss JN, Karma A, MacLellan WR, Deng M, Rau CD, Rees CM, Wang J, Wisniewski N, Eskin E, Horvath S, Qu Z, Wang Y, Lusis AJ . "Good enough solutions" and the genetics of complex diseases. Circ Res 2012; 111: 493–504.
Niu W, Wu S, Zhang Y, Li W, Ji K, Gao P, Zhu D . Validation of genetic association in apelin-AGTRL1 system with hypertension in a larger Han Chinese population. J Hypertens 2010; 28: 1854–1861.
Niu W, Qi Y . An updated meta-analysis of endothelial nitric oxide synthase gene: three well-characterized polymorphisms with hypertension. PLoS One 2011; 6: e24266.
Niu W, Qi Y, Qian Y, Gao P, Zhu D . The relationship between apolipoprotein E epsilon2/epsilon3/epsilon4 polymorphisms and hypertension: a meta-analysis of six studies comprising 1812 cases and 1762 controls. Hypertens Res 2009; 32: 1060–1066.
Marteau JB, Zaiou M, Siest G, Visvikis-Siest S . Genetic determinants of blood pressure regulation. J Hypertens 2005; 23: 2127–2143.
Zhao L, Stamler J, Yan LL, Zhou B, Wu Y, Liu K, Daviglus ML, Dennis BH, Elliott P, Ueshima H, Yang J, Zhu L, Guo D . Blood pressure differences between northern and southern Chinese: role of dietary factors: the International Study on Macronutrients and Blood Pressure. Hypertension 2004; 43: 1332–1337.
Niu W, Qi Y . Association of alpha-adducin and G-protein beta3 genetic polymorphisms with hypertension: a meta-analysis of Chinese populations. PLoS One 2011; 6: e17052.
Kessler T, Schunkert H . Clinical validation of genetic markers for improved risk estimation. Eur J Prev Cardiol 2012; 19 (2 Suppl): 25–32.
Gu M, Dong X, Zhang X, Wang X, Qi Y, Yu J, Niu W . Strong association between two polymorphisms on 15q25.1 and lung cancer risk: a meta-analysis. PLoS One 2012; 7: e37970.
Jin W, Su X, Xu M, Liu Y, Shi J, Lu L, Niu W . Interactive association of five candidate polymorphisms in Apelin/APJ pathway with coronary artery disease among Chinese hypertensive patients. PLoS One 2012; 7: e51123.
Ihsan R, Chauhan PS, Mishra AK, Yadav DS, Kaushal M, Sharma JD, Zomawia E, Verma Y, Kapur S, Saxena S . Multiple analytical approaches reveal distinct gene-environment interactions in smokers and non smokers in lung cancer. PLoS One 2011; 6: e29431.
Acknowledgements
This study received grants from the Shanghai Rising Star Program (11QA1405500), the Beijing New Star Program (Z111107054511072) and the National Natural Science Foundation of China (30900808, 81000109).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Hypertension Research website
Supplementary information
Rights and permissions
About this article
Cite this article
Qi, Y., Zhao, H., Wang, Y. et al. Replication of the top 10 most significant polymorphisms from a large blood pressure genome-wide association study of northeastern Han Chinese East Asians. Hypertens Res 37, 134–138 (2014). https://doi.org/10.1038/hr.2013.132
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2013.132
Keywords
This article is cited by
-
A novel predicted model for hypertension based on a large cross-sectional study
Scientific Reports (2020)
-
Common variant rs11191548 near the CYP17A1 gene is associated with hypertension and the serum 25(OH) D levels in Han Chinese
Journal of Human Genetics (2018)
-
A Review of the Genetics of Hypertension with a Focus on Gene-Environment Interactions
Current Hypertension Reports (2017)
-
The rs3754777 polymorphism of the STK39 gene is associated with essential hypertension in central south Chinese Han males
Hypertension Research (2016)
-
Association between a polymorphic poly-T repeat sequence in the promoter of the somatostatin gene and hypertension
Hypertension Research (2016)
