
Abstract
Hypertension is one of the common diseases in the elderly. The prevalence of hypertension markedly increases with advancing age. Both aging and hypertension have a critical role in cardiovascular and cerebrovascular complications. Although aging and hypertension, either independently or collectively, impair endothelial function, aging and hypertension may have similar cascades for the pathogenesis and development of endothelial dysfunction. Nitric oxide (NO) has an important role in regulation of vascular tone. Decrease in NO bioavailability by endothelial dysfunction would lead to elevation of blood pressure. An imbalance of reduced production of NO or increased production of reactive oxygen species, mainly superoxide, may promote endothelial dysfunction. One possible mechanism by which the prevalence of hypertension is increased in relation to aging may be advancing endothelial dysfunction associated with aging through an increase in oxidative stress. In addition, endothelial cell senescence is also involved in aging-related endothelial dysfunction. In this review, we focus on recent findings and interactions between endothelial function, oxidative stress and hypertension in aging.
Similar content being viewed by others

Introduction
Hypertension causes fatal cardiovascular diseases as a silent killer. It is well known that hypertension is one of the common diseases in the elderly. The prevalence of hypertension markedly increases with advancing age. Both aging and hypertension are independent predictors of cardiovascular and cerebrovascular outcomes.1, 2 Aging is associated with alterations of vascular structure and function through various pathways, including oxidative stress, cell senescence and inflammation.3, 4, 5 In addition, the presence of hypertension accelerates aging-related alterations of vascular structure and function, particularly endothelial function.6, 7
Endothelial dysfunction is an early feature of atherosclerosis and vascular diseases in humans.8 A great number of studies have shown that aging and hypertension are associated with impairment of endothelium-dependent vascular relaxation in coronary, forearm and renal arteries, and endothelial dysfunction, which is involved in the development of atherosclerosis, was found to increase the risk of cardiovascular and cerebrovascular diseases.9, 10, 11, 12, 13, 14, 15, 16, 17 It is expected that improvement or augmentation of endothelial function will prevent the development of atherosclerosis, resulting in reduction in cardiovascular and cerebrovascular events.
Several possible mechanisms by which advanced aging and hypertension impair endothelial function have been postulated. An imbalance of reduced production of nitric oxide (NO) or increased production of reactive oxygen species (ROS), mainly superoxide, may promote endothelial dysfunction.18, 19, 20 The key mechanism by which endothelium-dependent vasodilation is impaired is an increase in oxidative stress that inactivates NO. When considering aging-related endothelial dysfunction, the role of endothelial cell senescence in endothelial dysfunction should also be discussed.
In this review, we focus on recent findings and putative mechanisms by which aging, aging-associated hypertension and hypertension per se, either independently or collectively impair endothelial function.
Endothelial function
Until 1981, it was thought that the vascular endothelium functions as a wall separating the vessel wall and the inside cavity. If the endothelium of the whole body can be collected, its total weight would be equal to the liver, its total area would be equal to six tennis courts and its total length would be equal to two and half times around the globe, 100 000 km (Figure 1).21 In addition, it is well known that the endothelium secretes various vasoactive agents, such as the vasodilators NO, prostacyclin and endothelium-derived hyperpolarizing factor and the vasoconstrictors, such as endothelin-1, angiotensin II (Ang II) and thromboxane A2.22, 23, 24, 25 It is concluded that the endothelium is the biggest endocrine organ in the human body. A healthy endothelium maintains vascular tone and structure by regulating the balance between vasodilation and vasoconstriction, growth inhibition and growth promotion, antithrombosis and prothrombosis, anti-inflammation and proinflammation, and also antioxidation and pro-oxidation.23, 24, 25, 26 The simple molecule NO regulates basal vascular tone, at least in part, by about 50% in the brachial artery in humans.11, 26 Thus, loss of healthy endothelial function becomes a trigger of atherosclerosis. Endothelial dysfunction is the initial step in the pathogenesis of arteriosclerosis, resulting in cardiovascular complications.8 Indeed, the cumulative cardiovascular event rates in hypertensive patients with high-grade endothelial dysfunction were higher than in hypertensive patients with low-grade endothelial dysfunction.27 These findings suggest that forearm endothelial dysfunction is a prediction of future cardiovascular events in patients with hypertension. Lerman and Zeiher28 reported the results of multivariant analysis of hazard ratios of studies showing an association between coronary or peripheral endothelial function and cardiovascular events. In cardiovascular diseases other than hypertension, endothelial dysfunction is strongly and independently associated with cardiovascular events.29, 30 Hypertension, diabetes mellitus, dyslipidemia, aging, smoking, obesity and menopause are contributing risk factors in cardiovascular and cerebrovascular disease. These diseases and classical cardiovascular risk factors are associated with endothelial dysfunction. It is thought that endothelial function is a therapeutic target for atherosclerosis. Endothelial function is restored by appropriate interventions, including pharmacological therapy, such as renin-angiotensin system inhibitors and statins, supplementation therapy and lifestyle modifications.11, 31, 32, 33, 34, 35

Structure and function of endothelial cells and putative process from endothelial dysfunction to cardiovascular complications (modified by Higashi et al.)21 MI indicates myocardial infarction.
Assessment of endothelial function
In experimental studies, methods for assessment of endothelial function have been established by using a ring experiment protocol, endothelial functional alteration in expression of transcriptional factors and genes and genetic ablation of endothelial NO synthase (eNOS) in animal models.36, 37 It is clinically important to estimate the degree of endothelial dysfunction. Several methods have been used to assess endothelial function in humans. However, unfortunately, there is no gold standard for assessing endothelial function. Recently, several investigators, including us, have evaluated the effects of intra-arterial infusion of NO agonists, such as acetylcholine, methacholine and bradykinin, and intra-arterial infusion of NO antagonists on forearm blood flow using a mercury-filled Silastic strain-gauge plethysmography and the effects on coronary blood flow using a Doppler flow guide wire.9, 10, 11, 17, 26, 27, 35 The responses to intra-arterial infusion of vasoactive agents should be most suitable for assessing endothelial function, because the use of agonists to stimulate NO release and the use of antagonists of NO allow us to draw more specific conclusions concerning the role of basal and stimulated NO release. However, the invasive methods are time consuming and are a burden for patients. A noninvasive method for measuring forearm blood flow response to reactive hyperemia would also be useful for assessing endothelial function.38, 39 Measurement of flow-mediated vasodilation (FMD) in the brachial artery using ultrasound is noninvasive and reflects NO production very well.40, 41 It is accepted that measurement of forearm blood flow responses to vasoactive agents and reactive hyperemia is an index of resistance artery endothelial function and that measurement of FMD is an index of conduit artery endothelial function. Recently, finger plethysmography peripheral arterial tonometry has also been used.42, 43 Circulating levels of nitrite/nitrate, cyclic guanosine 3′,5′-monophosphate, vascular cell adhesion molecule-1, intracellular adhesion molecule-1, monocyte chemoattractant protein-1, plasminogen activator inhibitor-1, von Willebrand factor, asymmetrical dimethylarginine, endothelial microparticles and progenitor cells are also measured as indices of endothelial function.21, 44, 45, 46, 47 However, these measurements do not directly reflect the production of NO from endothelial cells and are not ‘function’. Measurement of circulating levels of NO metabolites, inflammatory markers and adhesion molecules should be used as an adjuvant to the measurement of forearm blood flow responses to vasoactive agents or FMD.
Endothelial dysfunction and aging
Aging may alter the structure and function of vascular components, such as the endothelium, intimas and smooth muscle cells, resulting in an increase in the risk of development of cardiovascular and cerebrovascular diseases, which are related to hypertension.3, 4, 5 In particular, aging-related endothelial dysfunction would contribute to pathogenesis, maintenance and development of atherosclerosis. Endothelial dysfunction is the initial step of atherosclerosis and is involved in the development of atherosclerosis. Attenuation of endothelium-dependent vasodilation has been observed in elderly subjects and animal models.48, 49
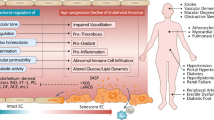
Several possible mechanisms by which advancing age impairs endothelial function are postulated (Figure 2). An imbalance between NO and ROS, so-called ‘oxidative stress,’ should be a key regulator of age-induced endothelial dysfunction. Aging activates nicotinamide-adenine dinucleotide phosphate (NADPH) oxidase, xantine oxidase, cyclooxygenase and mitochondrial electron transport and inactivates the antioxidant system, including superoxide dismutase (SOD), glutathione peroxidase (GPx) and catalase, leading to an increase in ROS production and decrease in ROS degradation. First, ROS directly inhibit NO activity. ROS activate the PI3K/Ras/Akt/MAPK pathway, related to redox transcriptional factors, leading to redox gene expression, which results in inhibition of eNOS mRNA expression and eNOS activity. Oxidation of tetrahydrobiopterin (BH4) by ROS induces eNOS uncoupling, and eNOS uncoupling produces ROS rather than NO.50 Under the condition of oxidative stress, BH4 predominantly produces superoxide, leading to peroxinitrite.51 Recently, the role of endothelial progenitor cells (EPCs) in endothelial function has been noted. Hill et al.44 have shown that the number of EPCs is decreased in relation to the cumulative number of Framingham risk factors and that the number of EPCs is correlated with endothelial function measured by FMD. In the early stage of endothelial dysfunction, impaired endothelial cells should be repaired by bone-marrow-derived EPCs. Decrease in the number of EPCs in bone marrow or inhibition of EPC mobilization may contribute to progression of endothelial dysfunction. Werner et al.52 reported that the cumulative event-free survival in analysis of a first major cardiovascular event at 12 months, according to levels of circulating EPCs at the time of enrollment in patients with cardiovascular disease. The cumulative event-free survival rate increased stepwise across three increasing baseline levels of EPCs in an analysis of death from cardiovascular causes. These finding suggest that the number of EPCs is a predictor of cardiovascular events. In addition, several investigators, including us, have shown a significant relationship between the member of EPCs and endothelial function in patients with cardiovascular diseases and even in normal subjects.44, 45 In our study, the number of EPCs significantly correlated with the number of total risk factors. Multiple regression analysis showed that age and hypertension are independent predictors of the number of EPCs.45 Experimental and clinical studies have clearly shown that excessive oxidative stress decreases the number of EPCs and impairs EPCs function.53, 54 Under the condition of excessive oxidative stress, aging-associated decrease in NO bioavailability and decrease in the number of EPCs and EPC mobilization may form a vicious circle and contribute to endothelial dysfunction.

Putative mechanisms by which advancing age and hypertension impairs endothelial function. BH4, tetrahydrobiopterin; eNOS, endothelial nitric oxide synthase; EPC, endothelial progenitor cell; NADPH, nicotinamide-adenine dinucleotide phosphate; NO, nitric oxide; ROS, reactive oxygen species; SOD superoxide dismutase.
Finally, several lines of evidence have shown that eNOS activity and NO production are decreased in senescent endothelial cells.55, 56 The process of endothelial cell senescence may have a critical role in endothelial dysfunction associated with aging (Figure 3).

Putative process of endothelial cell senescence with aging and putative process from endothelial cell senescence to endothelial dysfunction. eNOS, endothelial nitric oxide synthase; ROS indicates reactive oxygen species; TERT, telomerase reverse transcriptase.
Endothelial dysfunction and hypertension
Several investigators have clearly shown that endothelial function is impaired in animal models of hypertension.36 Systolic blood pressure was elevated by approximately 30 mm Hg in eNOS knockout mice compared with that in wild-type mice.37 In hypertensive patients also, endothelium-dependent vascular relaxation in coronary, forearm and renal arteries was found to be impaired, and endothelial dysfunction, which is involved in the development of atherosclerosis, was found to increase the risk of cardiovascular and cerebrovascular diseases.9, 10, 11, 12, 13, 14, 15, 27, 35 As Panza et al.9 reported for the first time in 1990 that the dose–response curve obtained by acetylcholine in patients with hypertension was smaller than the curve in normal controls and that the dose–response curves obtained by sodium nitropreside were similar in the two groups, suggesting that endothelial function, but not smooth muscle function, is selectively impaired in patients with hypertension, a large number of studies have shown that hypertension is associated with endothelial dysfunction.
However, although various factors contribute to the impartment of endothelial function in hypertension, the precise mechanisms remain unclear. Initially, agonists bind to receptors and/or shear stress activates eNOS, and NO, which is produced by L-arginine in the presence of eNOS in the endothelium, activates cytosolic guanylate cyclase and increases cGMP content in vascular smooth muscle cells, resulting in relaxation of vascular tone. Thus, it seems reasonable to assume that there is a problem somewhere in this L-arginine-NO-cGMP pathway. Several investigators have reported possible mechanisms of impairment of endothelial function in hypertension: increase in amount of the endogenous eNOS inhibitor asymmetrical dimethylarginine, increases in amounts of vasoconstrictors, such as Ang II, endothelin-1 and norepinephrine, and inactivation of NO by ROS.57, 58
Several studies using animal models of hypertension and patients with hypertension have shown that endothelial dysfunction is associated with an increase in ROS.58, 59 Amounts of antioxidant scavengers, such as SOD, GPx, catalase and vitamin C and E, are decreased in patients with hypertension.60 NADPH oxidase, which is a major source of production of ROS in vessel walls, is activated in hypertensive rats.61, 62 It has also been shown that ascorbic acid restores impaired endothelium-dependent vasodilation in patients with essential hypertension.63 Therefore, enhanced production of ROS and an attenuated antioxidant system may contribute to endothelial dysfunction in patients with hypertension. Enhanced NO inactivation caused by excess ROS production, rather than decreased NO production, may have an important role in impairment of endothelium-dependent vasodilation. These findings suggest that a decrease in NO inactivation contributes to the improvement in endothelial dysfunction in patients with hypertension. In various pathophysiological states, including hypertension, xanthine oxidase, NADPH oxidase and uncoupled eNOS are likely enzymatic sources contributing to increased production of ROS.64 An imbalance of decreased production of NO or increased production of ROS promotes endothelial dysfunction, leading to remodeling, platelet aggregation, loss of vasodilation, inflammation and smooth muscle cell growth.
Moreover, there is the question of whether endothelial dysfunction is a cause or consequence of hypertension. Several lines of evidence have suggested that endothelial function is impaired as blood pressure increases and that the degree of dysfunction is related to the magnitude of blood pressure elevation,64, 65 indicating that endothelial dysfunction is a consequence of hypertension. In contrast, Taddei et al.66 recently presented interesting results showing that young offspring of essential hypertensive patients who have a family history of hypertension are characterized by a reduced response to acetylcholine linked to a defect in the NO pathway, suggesting that endothelial dysfunction may be a cause of hypertension. There is still insufficient evidence for a conclusion to be reached.
Oxidative stress
NADPH oxidase
NADPH oxidase is the most important source of ROS in the vasculature.67 NADPH oxidase is a multisubunit complex composed of cytosolic components, such as p47phox, p67phox and Rac 1, and membrane-spanning components, such as p22phox and gp91phox. The production of ROS by activated NADPH oxidase is mediated by several pathways.59, 60, 68 Ang II-induced NADPH oxidase activation is one of the major sources of ROS in atherosclerosis.18, 19, 67, 68 Zalba et al.68 showed that endothelial dysfunction is due to an excess of ROS rather than a decrease in NO production in the aorta of spontaneously hypertensive rats and is associated with both upregulation of p22phox mRNA expression and increased activity of NADPH oxidase. Upregulation of p22phox mRNA expression is a key component of Ang II-induced NADPH oxidase activation, and increased expression levels of other components also have an important role in this oxidase under pathological conditions.18 Increased mRNA expression levels of p47phox, p67phox, p22phox and gp91phox have been found in internal mammary arteries of patients with cardiovascular diseases and diabetes mellitus. Guzik et al.19 reported that impairment of acetylcholine-induced vasodilation, increase in NADPH oxidase activity and NADPH oxidase-induced ROS generation in saphenous veins in patients with cardiovascular diseases are related to each other and are associated with increased risk of atherosclerosis. In addition, patients with renovascular hypertension are ideal models for determining how endothelium-dependent vasodilation is affected by excess Ang II and Ang II-related increase in oxidative stress.17 Renal angioplasty decreased plasma renin activity, plasma Ang II concentration and serum malondialdehyde-modified low-density lipoprotein (MDA-LDL) concentration and urinary 8-hydroxy-2′-deoxyguanosine (8-OHdG) excretion, indices of oxidative stress, in patients with renovascular hypertension. After renal angioplasty, forearm blood flow response to acetylcholine was enhanced in patients with renovascular hypertension. Co-infusion of the antioxidant vitamin C augmented the forearm blood flow response to acetylcholine before angioplasty but not after angioplasty. The increase in maximal forearm blood flow response to acetylcholine correlated with the decrease in urinary excretion of 8-OHdG and the decrease in serum concentration of MDA-LDL. These findings suggest that endothelial function is impaired in relation to the severity of oxidative stress in humans.
SOD, GPx and catalase
Protective antioxidant mechanisms are complex and multifactorial. The antioxidant defense system, including SOD, GPx and catalase, scavenges ROS in the vasculature, resulting in inhibition of NO degradation. The susceptibility of vascular cells to oxidative stress is a function of the overall balance between the degree of oxidative stress and the antioxidant defense capability. The antioxidant enzyme SOD rapidly dismutates superoxide to hydrogen peroxide. SOD has been identified as three enzymatic types: Cu/Zn SOD, Mn SOD and extracellular SOD. Destruction of the antioxidant system, including decreased antioxidant enzyme activity and ROS scavenging ability, may contribute to oxidative stress in patients with atherosclerosis. Indeed, circulating levels of SOD diminished in relation to aging.69 Various interventions such as administration of antioxidant vitamins and antihypertensive agents and exercise training have been shown to enhance the protein levels and enzymatic activities of SODs, such as Cu/Zn SOD and Mn SOD, in the vascular endothelium and smooth muscle cells of the aorta in various animal models.70, 71 In the vasculature of humans, approximately 50% of total SOD is extracellular SOD.72 Fukai et al.73 demonstrated that exercise for 3 weeks increased eNOS and extracellular SOD protein levels in wild-type mice but had no effect on extracellular SOD protein levels in eNOS-knockout mice and that the effect of endothelium-derived NO on extracellular SOD protein level is mediated by the cGMP/protein kinase G-dependent pathway. Taken together, impairment of the antioxidant defense system associated with aging and hypertension may contribute to endothelial dysfunction.
Tetrahydrobiopterin (BH4)
eNOS requires several cofactors, such as heme, flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN), as well as BH4 for elicitation of full enzymatic activity.74, 75 BH4 is an allosteric factor in the coupling of the oxidase and reductase domains of eNOS.50 Recently, it has been reported that a deficiency of BH4 results in a decrease in NO synthesis and an increase in production of ROS.51 Sowers20 emphasized the role of BH4 in oxidative stress and endothelial function. In animals and humans of advanced age, dysfunctional eNOS with insufficient BH4 induces production of ROS, resulting in a decrease in NO bioavailability.75 Reduced availability of BH4 may contribute to the development and maintenance of atherosclerosis. In addition, it has been demonstrated that supplementation of BH4 or folic acid improves endothelial function in smokers, in patients with diabetes, hypertension, hypercholesterolemia and chronic heart failure and in elderly subjects.76, 77, 78, 79, 80, 81, 82 Co-infusion of BH4 resulted in a significant increase in acetylcholine-induced vasodilation in healthy men.78 Aging was found to be significantly correlated with acetylcholine-induced vasodilation, urinary 8-OHdG, serum MDA-LDL and change in acetylcholine-induced vasodilation after co-infusion of BH4. Infusion of NG-monomethyl-L-arginine, an NO synthase inhibitor, abolished the BH4-induced enhancement of forearm vasorelaxation evoked by acetylcholine. Also in hypertension, a deficiency of BH4 decreases NO synthesis and increases superoxide production, and reduced availability of BH4 may contribute to the development and maintenance of hypertension. Indeed, during co-infusion of BH4, the forearm blood flow response to acetylcholine in hypertensive patients increased significantly to the level of normal controls.78 After NG-monomethyl-L-arginine infusion, BH4-induced augmentation of endothelium-dependent vasodilation was completely abolished. These findings suggest that a deficiency of BH4 may be involved in the pathogenesis of disturbances in endothelium-dependent vasodilation related to aging and hypertension through decrease in NO production and increase in oxidative stress.
Several lines of evidence have suggested that oxidative DNA damage is increased in relation to aging.83, 84 Levels of aldehydes, such as MDA-LDL and 4-hydroxy-2-nonenal (4-HNE), have been proposed as a biological signature of clinical in vivo LDL oxidation.85, 86, 87, 88, 89 Mutlu-Turkoglu et al.87 reported that plasma MDA levels were increased in elderly subjects compared with those in young subjects. Lovell et al.89 have shown that ventricular fluid levels of 4-HNE are significantly higher in patients with Alzheimer's disease, an age-related disease, than in control subjects. We also confirmed that aging correlated with urinary 8-ODdG excretion and serum MDA-LDL concentration, suggesting that oxidative stress progressively increases with aging.79 Several investigators have shown that both oxidized LDL and native LDL downregulate eNOS mRNA and protein levels in endothelial cells.90, 91 Interestingly, 4-HNE mediated eNOS uncoupling and superoxide generation through alteration of BH4 homeostasis in bovine aorta endothelial cells.92 Therefore, although the effect of MDL-LDL on endothelial function in humans is not clear, MDA-LDL is not merely a marker of oxidative stress but also may directly impair endothelial function through a decrease in the expression of eNOS.
While eNOS activation at optimal levels of BH4 leads to production of NO, activation of NOS in the absence of or in the presence of decreased levels of BH4 results in generation of superoxide rather than NO in the vascular endothelium.16, 17 Under conditions of BH4 deficiency, electron flow from the reductase domain to the oxidase domain is diverted to molecular oxygen rather than to L-arginine, a substrate of NO, leading to eNOS uncoupling. In this condition, uncoupled eNOS produces superoxide rather than NO.20 In aging models, dysfunctional eNOS with insufficient BH4 causes superoxide generation, resulting in decreased NO bioaviliability.75 It has recently been reported that degradation of BH4 by ROS, including peroxynitrite, superoxide and hydrogen peroxide, is associated with downregulation of eNOS.93 These findings suggest that BH4 deficiency and decreased eNOS activity cause endothelial dysfunction in elderly subjects and in patients with hypertension through an increase in oxidative stress. Therefore, BH4 supplementation may increase the intracellular content of BH4 and augment acetylcholine-induced vasodilation by inhibition of NO inactivation.
RhoA/Rho-associated kinase (ROCK)
Recent studies have shown that the ROCK family, one of the several small GTPase Rho effectors, has major roles in actin cytoskeleton organization,94 smooth muscle contraction95 and gene expression.96 Results of previous studies have shown that ROCK has a role in the contraction of vascular smooth muscle cells. ROCK activates myosin light-chain kinase by phosphorylation of the myosin-binding subunit in myosin light-chain phosphatase, leading to contraction of vascular smooth muscle cells.97, 98 Indeed, the RhoA/ROCK pathway has been shown to be involved in the formation of atherosclerotic lesions, vasoconstriction and myocardial hypertrophy and to be activated in patients with hypertension and in patients with coronary artery disease.99, 100, 101, 102, 103, 104 Recently, we have shown that FMD is an independent predictor of leukocyte ROCK activity in healthy men and men with cardiovascular risk factors but without established cardiovascular or cerebrovascular diseases, suggesting that cumulative cardiovascular risk, including aging and hypertension, may enhance ROCK activity and endothelial dysfunction.105 Activation of the RhoA/ROCK pathway impairs NO bioavailability through inhibition of eNOS mRNA stability, eNOS posphorylation at Ser 1177 and the Akt/PI3K pathway and enhancement of eNOS phosphorylation at Thr495.106, 107, 108 Several investigators have shown an interaction between the RhoA/ROCk pathway and ROS.109, 110 Indeed, ROS induced by hyperglycemia enhances ROCK activity, leading to atherothrombogenesis through an increase in expression of plasminogen activator inhibitor-1 in vascular endothelial cells.111 It is well known that cigarette smoking decreases NO bioavailability through the production of ROS. Several investigators, including us, have demonstrated that there is a possible association of ROCK activity with oxidative stress and that smoking enhances the activation of ROCKs in vascular smooth muscle cells in vivo and in vitro.98, 109, 112 These findings suggest a correlation between oxidative stress and ROCK activity in humans. Hernandez-Perera et al.113 showed that Rho is required for the basal expression of preproendothelin-1 in vascular endothelial cells, which gives rise to endothelin-1. On the other hand, several investigators have shown that NO blocks the migration of RhoA from the cytosol to the membrane, thereby inhibiting the RhoA/ROCK signaling pathway.114, 115 It is thought that there is an interaction between activated ROCK and endothelial dysfunction in human vessels. Indeed, we have confirmed that hypertension is associated with both endothelial dysfunction and increased ROCK activity, and that endothelial dysfunction and increased ROCK activity are simultaneously improved by an aldosterone blocker in patients with hypertension. These findings indicate close interactions between ROCK activity, endogenous NO and oxidative stress.
Interestingly, ROCK activity was significantly correlated with systolic blood pressure even in healthy young male subjects with normal blood pressure.116 This finding is supported by results of previous studies showing that ROCKs are involved in the pathogenesis of hypertension through, at least in part, causing vascular smooth muscle cell contraction.117, 118, 119 There is a possibility that ROCK activity regulates blood pressure from the normal range to high levels.
Endothelial cell senescence
If endothelial cells are altered to senescence, structural and functional changes in endothelial cells, including alteration in gene expression occurs in relation to the advancing cell senescence. Telomeres are located at the ends of the chromosome to protect DNA damage signals and to diminish the activation of DNA repair in various types of cells, including endothelial cells.120, 121 Activity of telomerase, a catalytic subunit telomerase reverse transcriptase (TERT), regulates the DNA length of telomeres.122, 123 Therefore, telomere length and activity of telomerase, especially TERT, have a critical role in endothelial cell senescence. It is well known that aging and hypertension are associated with shortening of telomeres induced by decrease in TERT activity in endothelial cells.124, 125, 126 However, unfortunately, it remains unclear whether telomere shortening is a cause or consequence of aging.
Endothelial cell senescence per se can cause endothelial dysfunction. Several lines of evidence have shown that telomere length in endothelial cells from human aortas and arteries shortens in relation to aging.127, 128 Decreases in both NO production and eNOS activity in human umbilical vein endothelial cells and human aortic endothelial cells (HAECs) are associated with aging.129, 130 Matsushita et al.130 demonstrated that shear stress-induced increase in NO production was diminished in senescent HAECs and that stable expression of TERT restored the decreased eNOS expression and NO production associated with aging. Minamino et al.131 reported that HAECs from human atherosclerotic lesions presented phenotypes of senescent cells and decreases in eNOS expression and eNOS activity. Introduction of TERT extended the life span and restored endothelial functional alteration associated with senescence in HAECs. Interestingly, exogenous NO reduced human umbilical vein endothelial cell senescence and delayed age-dependent inhibition of telomerase activity, suggesting that telomerase inactivation precedes endothelial cell senescence and that NO prevents age-related downregulation of telomerase activity and delays endothelial cell senescence.132 Nakashima et al.133 showed by measuring the mean telomere restriction fragment length that telomere lengths in white blood cells shorten in parallel to a decline in endothelial function by FMD in patients with various degrees of cardiovascular damage as well as in healthy subjects.
The process of endothelial dysfunction associated with aging in relation to endothelial cell senescence is postulated as follows (Figure 3): Aging and hypertension activate NADPH oxidase, xantine oxidase, cyclooxygenase and mitochondrial electron transport and inactivate the antioxidant system, leading to increase in ROS production and decrease in ROS degradation. ROS induce export of nuclear TERT into the cytosol through activation of Src-family kinases.134, 135 Inhibition of nuclear TERT activity shortens telomere length, leading to decrease in the endothelial cell’s life span, alteration in gene expression and change from phenotype of young cells to phenotype of old cells.136 In addition, telomere shortening stimulates activation of p53, p21 and p16 proteins, which are triggers of cell senescence.137, 138 The endogenous eNOS inhibitor asymmetrical dimethylarginine also accelerates endothelial cell senescence through increase in production of ROS and inhibition of NO production.139 An imbalance between NO and ROS may be the initial step of endothelial cell senescence and may have an important role in endothelial cell senescence. Endothelial cell senescence results in endothelial dysfunction through various pathways.
Conclusions
Reduced NO bioavailability and reduced number and function of EPCs by ROS and endothelial cell senescence may contribute to endothelial dysfunction in aging. In hypertension also, similar mechanisms may work in the process of endothelial dysfunction. Aging, aging-associated hypertension and hypertension per se, either independently or collectively, impair endothelial function, leading to atherosclerosis, resulting in cardiovascular and cerebrovascular outcomes. It is expected that improvement or augmentation of endothelial function will prevent the development of atherosclerosis, resulting in reduction in cardiovascular and cerebrovascular events. Intervention to reduce oxidative stress should be an effective strategy for treatment of atherosclerosis, including aging and hypertension, through, at least in part, improvement in endothelial function and endothelial cell senescence.
References
Kannel WB, Gordon T The Framingham Study: An epidemiological investigation of cardiovascular disease. Section 30. Some characteristics related to the incidence of cardiovascular disease and death: The Framingham Study 18-year Follow-up. US Dept of Health, Education, and Welfare Publication No. (NIH) 74-599 US Government Printing Office: Washington, DC. 1974.
Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Roger VL, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J, American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation 2010; 121: e46–e215.
Wei JY . Age and the cardiovascular system. N Engl J Med 1992; 327: 1735–1739.
Folkow B, Svanborg A . Physiology of cardiovascular aging. Physiol Rev 1993; 73: 725–764.
Butt HZ, Atturu G, London NJ, Sayers RD, Bown MJ . Telomere length dynamics in vascular disease: a review. Eur J Vasc Endovasc Surg 2010; 40: 17–26.
Higashi Y, Oshima T, Ozono R, Watanabe M, Matsuura H, Kambe M, Kajiyama G . Aging and severity of hypertension attenuate endothelialum-dependent renal vascular relaxation in humans. Hypertension 1997; 30: 252–258.
Kimura Y, Matsumoto M, Den YB, Iwai K, Munehira J, Hattori H, Hoshino T, Yamada K, Kawanishi K, Tsuchiya H . Impaired endothelial function in hypertensive elderly patients evaluated by high resolution ultrasonography. Can J Cardiol 1999; 15: 563–568.
Ross R . Atherosclerosis--an inflammatory disease. N Engl J Med 1999; 340: 115–126.
Panza JA, Quyyumi AA, Brush JE, Epstein SE . Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N Engl J Med 1990; 323: 22–27.
Linder L, Kiowski W, Buhler FR, Lüscher TF . Indirect evidence for release of endothelium-derived relaxing factor in human forearm circulation in vivo: blunted response in essential hypertension. Circulation 1990; 81: 1762–1767.
Higashi Y, Sasaki S, Kurisu S, Yoshimizu A, Sasaki N, Matsuura H, Kajiyama G, Oshima T . Regular aerobic exercise augments endothelium-dependent vascular relaxation in normotensive as well as hypertensive subjects: role of endothelium-derived nitric oxide. Circulation 1999; 100: 1194–1202.
Treasure CB, Klein JL, Vita JA, Manoukian SV, Renwick GH, Selwyn AP, Ganz P, Alexander RW . Hypertension and left ventricular hypertrophy are associated with impaired endothelium-mediated relaxation in human coronary resistance vessels. Circulation 1993; 87: 86–93.
Egashira K, Suzuki S, Hirooka Y, Kai H, Sugimachi M, Imaizumi T, Takeshita A . Impaired endothelium-dependent vasodilation in large epicardial and resistance coronary arteries in patients with essential hypertension: different responses to acetylcholine and substance P. Hypertension 1995; 25: 201–206.
Raij L . Nitric oxide and the kidney. Circulation 1993; 87: V26–V29.
Higashi Y, Oshima T, Ozono R, Watanabe M, Matsuura H, Kajiyama G . Effects of L-arginine infusion on renal hemodynamics in patients with mild essential hypertension. Hypertension 1995; 25: 898–902.
Creager MA, Cooke JP, Mendelsohn ME, Gallagher SJ, Coleman SM, Loscalzo J, Dzau VJ . Impaired vasodilation of forearm resistance vessels in hypercholesterolemic humans. J Clin Invest 1990; 86: 228–234.
Higashi Y, Sasaki S, Nakagawa K, Matsuura H, Oshima T, Chayama K . Endothelial function and oxidative stress in renovascular hypertension. N Engl J Med 2002; 346: 1954–1962.
Touyz RM . Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension 2004; 44: 248–252.
Guzik TJ, West NE, Black E, McDonald D, Ratnatunga C, Pillai R, Channon KM . Vascular superoxide production by NAD(P)H oxidase: association with endothelial dysfunction and clinical risk factors. Circ Res 2000; 86: E85–E90.
Sowers JR . Hypertension, angiotensin II, and oxidative stress. N Engl J Med 2002; 346: 1999–2001.
Higashi Y, Noma K, Yoshizumi M, Kihara Y . Oxidative stress and endothelial function in cardiovascular diseases. Circ J 2009; 73: 411–418.
Lüscher TF . Imbalance of endothelium-derived relaxing and contracting factors. Am J Hypertens 1990; 3: 317–330.
Vane JR, Anggard EE, Botting RM . Regulatory functions of the vascular endothelium. N Engl J Med 1990; 323: 27–36.
Vallance P, Collier J, Moncada S . Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet 1989; 2: 997–1000.
Vanhoutte PM . Endothelium and control of vascular function: state of the art lecture. Hypertension 1989; 13: 658–667.
Higashi Y, Nakagawa K, Kimura M, Noma K, Sasaki S, Hara K, Goto C, Oshima T, Chayama K, Yoshizumi M . Low body mass index is a risk factor for impaired endothelium-dependent vasodilation in humans: role of nitric oxide and oxidative stress. J Am Coll Cardiol 2003; 42: 256–263.
Perticone F, Ceravolo R, Pujia A, Ventura G, Iacopino S, Scozzafava A, Ferraro A, Chello M, Mastroroberto P, Verdecchia P, Schillaci G . Prognostic significance of endothelial dysfunction in hypertensive patients. Circulation 2001; 104: 191–196.
Lerman A, Zeiher AM . Endothelial function: cardiac events. Circulation 2005; 111: 363–368.
Suwaidi JA, Hamasaki S, Higano ST, Nishimura RA, Holmes DR, Lerman A . Long-term follow-up of patients with mild coronary artery disease and endothelial dysfunction. Circulation 2000; 101: 948–954.
Schachinger V, Britten MB, Zeiher AM . Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation 2000; 101: 1899–1906.
Ting HH, Timimi FK, Boles KS, Creager SJ, Ganz P, Creager MA . Vitamin C improves endothelium-dependent vasodilation in patients with non-insulin-dependent diabetes mellitus. J Clin Invest 1996; 97: 22–28.
McVeigh GE, Brennan GM, Johnston GD, McDermott BJ, McGrath LT, Henry WR, Andrews JW, Hayes JR . Impaired endothelium-dependent and independent vasodilation in patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 1992; 35: 771–776.
Treasure CB, Klein JL, Weintraub WS, Talley JD, Stillabower ME, Kosinski AS, Zhang J, Boccuzzi SJ, Cedarholm JC, Alexander RW . Beneficial effects of cholesterol-lowering therapy on the coronary endothelium in patients with coronary artery disease. N Engl J Med 1995; 332: 481–467.
Levine GN, Frei B, Koulouris SN, Gerhard MD, Keaney JF, Vita JA . Ascorbic acid reverses endothelial vasomotor dysfunction in patients with coronary artery disease. Circulation 1996; 93: 1107–1113.
Higashi Y, Sasaki S, Nakagawa K, Kurisu S, Yoshimizu A, Matsuura H, Kajiyama G, Oshima T . A comparison of angiotensin-converting enzyme inhibitors, calcium antagonists, beta-blockers, diuretics on reactive hyperemia in patients with essential hypertension: a multicenter study. J Am Coll Cardiol 2000; 35: 284–291.
Tsutsui M, Shimokawa H, Otsuji Y, Yanagihara N . Pathophysiological relevance of NO signaling in the cardiovascular system: novel insight from mice lacking all NO synthases. Pharmacol Ther 2010; 128: 499–508.
Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC . Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995; 377: 239–242.
Higashi Y, Sasaki S, Nakagawa K, Matsuura H, Kajiyama G, Oshima T . A noninvasive measurement of reactive hyperemia that can be used to assess resistance artery endothelial function in humans. Am J Cardiol 2001; 87: 121–125.
Higashi Y, Sasaki S, Nakagawa K, Matsuura H, Kajiyama G, Oshima T . Effect of angiotensin-converting enzyme inhibitor imidapril on reactive hyperemia in patients with essential hypertension - relationship between treatment periods and resistance artery endothelial function -. J Am Coll Cardiol 2001; 37: 863–870.
Benjamin EJ, Larson MG, Keyes MJ, Mitchell GF, Vasan RS, Keaney JF, Lehman BT, Fan S, Osypiuk E, Vita JA . Clinical correlates and heritability of flow-mediated dilation in the community: the Framingham Heart Study. Circulation 2004; 109: 613–619.
Fujimura N, Hata T, Soga J, Hidaka T, Idei N, Fujii Y, Mikami S, Maruhashi T, Iwamoto Y, Kihara Y, Chayama K, Kato H, Noma K, Liao JK, Higashi Y, . For ROCK investigator group. Selective mineralocorticoid receptor blocker eplerenone improves endothelial function and inhibits Rho-associated kinase activity in patients with essential hypertension. Clin Pharmacol Ther 2012; 91: 289–297.
Nohria A, Gerhard-Herman M, Creager MA, Hurley S, Mitra D, Ganz P . Role of nitric oxide in the regulation of digital pulse volume amplitude in humans. J Appl Physiol 2006; 101: 545–548.
Matsuzawa Y, Sugiyama S, Sugamura K, Nozaki T, Ohba K, Konishi M, Matsubara J, Sumida H, Kaikita K, Kojima S, Nagayoshi Y, Yamamuro M, Izumiya Y, Iwashita S, Matsui K, Jinnouchi H, Kimura K, Umemura S, Ogawa H . Digital assessment of endothelial function and ischemic heart disease in women. J Am Coll Cardiol 2010; 55: 1688–1696.
Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T . Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med 2003; 348: 593–600.
Umemura T, Soga J, Hidaka T, Takemoto H, Nakamura S, Jitsuiki D, Nishioka K, Goto C, Teragawa H, Yoshizumi M, Chayama K, Higashi Y . Aging and hypertension are independent risk factors for reduced number of circulating endothelial progenitor cells. Am J Hypertens 2008; 21: 1203–1209.
Böger RH, Bode-Böger SM, Szuba A, Tsao PS, Chan JR, Tangphao O, Blaschke TF, Cooke JP . Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation 1998; 98: 1842–1847.
Heiss C, Amabile N, Lee AC, Real WM, Schick SF, Lao D, Wong ML, Jahn S, Angeli FS, Minasi P, Springer ML, Hammond SK, Glantz SA, Grossman W, Balmes JR, Yeghiazarians Y . Brief secondhand smoke exposure depresses endothelial progenitor cells activity and endothelial function: sustained vascular injury and blunted nitric oxide production. J Am Coll Cardiol 2008; 51: 1760–1771.
Zeiher AM, Drexler H, Saurbier B, Just H . Endothelium-mediated coronary blood flow modulation in humans. Effects of age, atherosclerosis, hypercholesterolemia, and hypertension. J Clin Invest 1993; 92: 652–662.
Safar M, Chamiot-Clerc P, Dagher G, Renaud JF . Pulse pressure, endothelium function, and arterial stiffness in spontaneously hypertensive rats. Hypertension 2001; 38: 1416–1421.
Rodriguez-Crespo I, Gerber NC, Ortiz de Montellano PR . Endothelial nitric-oxide synthase. J Biol Chem 1996; 271: 11462–11467.
Kaufman S . New tetrahydrobiopterin-dependent systems. Annu Rev Nutr 1993; 13: 261–286.
Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, Böhm M, Nickenig G . Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med 2005; 353: 999–1007.
Murasawa S, Llevadot J, Silver M, Isner JM, Losordo DW, Asahara T . Constitutive human telomerase reverse transcriptase expression enhances regenerative properties of endothelial progenitor cells. Circulation 2002; 106: 1133–1139.
Ballard VL, Edelberg JM . Stem cells and regeneration of aging cardiovascular system. Circ Res 2007; 100: 1116–1127.
Hayashi T, Yano K, Matsui-Hirai H, Yokoo H, Hattori Y, Iguchi A . Nitric oxide and endothelial cellular senescence. Pharmacol Ther 2008; 120: 333–339.
Farsetti A, Grasselli A, Bacchetti S, Gaetano C, Capogrossi MC . The telomerase tale in vascular aging: regulation by estrogens and nitric oxide signaling. J Appl Physiol 2009; 106: 333–337.
Drexler H, Horning B . Endothelial dysfunction in human disease. J Mol Cell Cardiol 1999; 31: 51–60.
Cai H, Harrison DG . Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 2000; 87: 840–844.
Dijhorst-Oei LT, Stores ES, Koomans HA, Rabelink TJ . Acute stimultaneous stimulation of nitric oxide and oxygen radicals by angiotensin II in humans in vivo. J Cardiovasc Pharmacol 1999; 33: 420–424.
Irani K . Oxidant signaling in vascular cell growth, death and survival: a review of the roles of reactive oxygen species in smooth muscle and endothelail cell mitogenic and apoptotic signaling. Circ Res 2000; 87: 179–183.
Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG . Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. J Clin Invest 1996; 97: 1916–1923.
Hegde LG, Srivastava P, Kumari R, Dikshit M . Alterations in the vasoreactivity of hypertensive rat aortic rings: role of nitric oxide and superoxide radicals. Clin Exp Hypertens 1998; 20: 885–901.
Taddei S, Virdis A, Ghiadoni L, Magagna A, Salvetti A . Vitamin C improves endothelium-dependent vasodilation by restoring nitric oxide activity in essential hypertension. Circulation 1998; 97: 2222–2229.
Dohi Y, Thiel MA, Buhler FR, Lüscher TF . Activation of endothelial L-arginine pathway in resistance arteries. effect of age and hypertension. Hypertension 1990; 16: 170–179.
Hermann M, Flammer A, Lüscher TF . Nitric oxide in hypertension. J Clin Hypertens 2006; 8 (12 Suppl 4): 17–29.
Taddei S, Virdis A, Mattei P, Ghiadoni L, Sudano I, Salvetti A . Defective L-arginine-nitric oxide pathway in offspring of essential hypertensive patients. Circulation 1996; 94: 1298–1303.
Dzau VJ . Theodore Cooper Lecture: tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension 2001; 37: 1047–1052.
Zalba G, Beaumont FJ, San Jose G, Fortuno A, Fortuno MA, Etayo JC, Diez J . Vascular NADH/NADPH oxidase is involved in enhanced superoxide production in spontaneously hypertensive rats. Hypertension 2000; 35: 1055–1061.
Liu H, Yang Y, Huang G, Tan S, Liu Y . Positive association of pro-inflammatory biomarkers and increased oxidative stress in the healthy elderly. Arch Gerontol Geriatr 2012; 54: e8–e12.
Yamashita N, Hoshida S, Otsu K, Asahi M, Kuzuya T, Hori M . Exercise provides direct biphasic cardioprotection via manganese superoxide dismutase activation. J Exp Med 1999; 189: 1699–1706.
Rush JW, Turk JR, Laughlin MH . Exercise training regulates SOD-1 and oxidative stress in porcine aortic endothelium. Am J Physiol Heart Circ Physiol 2003; 284: H1378–H1387.
Stralin P, Karlsson K, Johansson BO, Marklund SL . The interstitium of the human arterial wall contains very large amounts of extracellular superoxide dismutase. Arterioscler Thromb Vasc Biol 1995; 11: 2032–2036.
Fukai T, Siegfried MR, Ushio-Fukai M, Cheng Y, Kojda G, Harrison DG . Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J Clin Invest 2000; 105: 1631–1639.
Hinzel B, John M, Klatt P, Bohem E, Mayer B . Ca2+/calmodulin-dependent formation of hydrogen peroxide by brain nitric oxide synthase. Biochem J 1992; 281: 627–630.
Chen EY, Kallwitz E, Leff SE, Cochran EJ, Mufson EJ, Kordower JH, Mandel RJ . Age-related decreases in GTP-cyclohydrolase-I immunoreactive neurons in the monkey and human substantia nigra. J Comp Neurol 2000; 426: 534–548.
Stoes E, Kastelein J, Cosentino F, Erkelens W, Wever R, Koomans H, Lüscher TF, Rabelink T . Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J Clin Invest 1997; 99: 41–46.
Setoguchi S, Hirooka Y, Eshima K, Shimokawa H, Takeshita A . Tetrahydrobiopterin improves impaired endothelium-dependent forearm vasodilation in patients with heart failure. J Cardiovasc Pharmacol 2003; 39: 363–368.
Higashi Y, Sasaki S, Nakagawa K, Fukuda Y, Matsuura H, Oshima T, Chayama K . Tetrahydrobiopterin improves impaired endothelium-dependent vasodilation in patients with essential hypertension. Am J Hypertens 2002; 15: 326–332.
Higashi Y, Sasaki S, Nakagawa K, Kimura M, Noma K, Hara K, Jitsuiki D, Goto C, Oshima T, Chayama K, Yoshizumi M . Tetrahydrobiopterin improves aging-related impairment of endothelium-dependent vasodilation through increase in nitric oxide production and decrease in reactive oxygen species. Atherosclerosis 2006; 186: 390–395.
Hayden MR, Tyagi SC . Is type 2 diabetes mellitus a vascular disease (atheroscleropathy) with hyperglycemia a late manifestation? The role of NOS, NO, and redox stress. Cardiovasc Diabetol 2003; 2: 1–10.
Moat SJ, McDowell IF . Homocysteine and endothelial function in human studies. Semin Vasc Med 2005; 5: 172–182.
Moreau KL, Meditz A, Deane KD, Kohrt WM . Tetrahydrobiopterin improves endothelial function and decreases arterial stiffness in estrogen-deficient postmenopausal women. Am J Physiol Heart Circ Physiol 2012; 302: H1211–H1218.
Fraga C, Shigenega M, Park J, Degan P, Ames BN . Oxidative damage to DNA during aging: 8-hydroxy-2′-deoxyguanosine in rat organ DNA and urine. Proc Natl Acad Sci USA 1990; 87: 4533–4537.
Leinonen J, Lehtimaki T, Toyokuni S, Okada K, Tanaka T, Hiai H, Ochi H, Laippala P, Rantalaiho V, Wirta O, Pasternack A, Alho H . New biomarker evidence of oxidative DNA damage in patients with non-insulin-dependent diabetes mellitus. FEBS Lett 1997; 417: 150–152.
Salonen JT, Yla-Herttuala S, Yamamoto R, Butler S, Korpela H, Salonen R, Nyyssönen K, Palinski W, Witztum JL . Autoantibody against oxidized LDL and progression of carotid atherosclerosis. Lancet 1992; 339: 883–887.
Palinski W, Miller E, Witztum JL . Immunization of low density lipoprotein (LDL) receptor-deficient rabbits with homologous malondialdehyde-modified LDL reduced atherogenesis. Proc Natl Acad Sci USA 1995; 92: 821–825.
Mutlu-Turkoglu U, Ilhan E, Oztezcan S, Kuru A, Aykac-Toker G, Uysal M . Age-related increases in plasma malondialdehyde and protein carbonyl levels and lymphocyte DNA damage in elderly subjects. Clin Biochem 2003; 36: 397–400.
Ma R, Yu J, Xu D, Yang L, Lin X, Zhao F, Bai F . Effect of felodipine with irbesartan or metoprolol on sexual function and oxidative stress in women with essential hypertension. J Hypertens 2012; 30: 210–216.
Lovell MA, Ehmann WD, Mattson MP, Markesbery WR . Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer's disease. Neurobiol Aging 1997; 18: 457–461.
Liao JK, Shin WS, Lee WY, Clark SL . Oxidized low-density lipoprotein decreases the expression of endothelial nitric oxide synthase. J Biol Chem 1995; 270: 319–324.
Vidal F, Colome C, Martinez-Gonzalez J, Badimon L . Atherogenic concentrations of native low-density lipoprotein down-regulate nitric-oxide-synthase mRNA and protein levels in endothelial cells. Eur J Biochem 1998; 252: 378–384.
Whitsett J, Picklo MJ, Vasquez-Vivar J . 4-Hydroxy-2-nonenal increases superoxide anion radical in endothelial cells via stimulated GTP cyclohydrolase proteasomal degradation. Arterioscler Thromb Vasc Biol 2007; 27: 2340–2347.
Kuzkaya N, Weissmann N, Harrison DG, Dikalov S . Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J Biol Chem 2003; 278: 22546–22554.
Amano M, Chihara K, Kimura K, Fukata Y, Nakamura N, Matsuura Y, Kaibuchi K . Formation of actin stress fibers and focal adhesions enhanced by Rho-kinase. Science 1997; 275: 1308–1311.
Kaibuchi K, Kuroda S, Amano M . Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu Rev Biochem 1999; 68: 459–486.
Chihara K, Amano M, Nakamura N, Yano T, Shibata M, Tokui T, Ichikawa H, Ikebe R, Ikebe M, Kaibuchi K . Cytoskeletal rearrangements and transcriptional activation of c-fos serum response element by Rho-kinase. J Biol Chem 1997; 272: 25121–25127.
Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K . Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 1996; 273: 245–248.
Kureishi Y, Kobayashi S, Amano M, Kimura K, Kanaide H, Nakano T, Kaibuchi K, Ito M . Rho-associated kinase directly induces smooth muscle contraction through myosin light chain phosphorylation. J Biol Chem 1997; 272: 12257–12260.
Mallat Z, Gojova A, Sauzeau V, Brun V, Silvestre JS, Esposito B, Merval R, Groux H, Loirand G, Tedgui A . Rho-associated protein kinase contributes to early atherosclerotic lesion formation in mice. Circ Res 2003; 93: 884–888.
Kataoka C, Egashira K, Inoue S, Takemoto M, Ni W, Koyanagi M, Kitamoto S, Usui M, Kaibuchi K, Shimokawa H, Takeshita A . Important role of Rho-kinase in the pathogenesis of cardiovascular inflammation and remodeling induced by long-term blockade of nitric oxide synthesis in rats. Hypertension 2002; 39: 245–250.
Rikitake Y, Kim HH, Huang Z, Seto M, Yano K, Asano T, Moskowitz MA, Liao JK . Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke 2005; 36: 2251–2257.
Rikitake Y, Oyama N, Wang CY, Noma K, Satoh M, Kim HH, Liao JK . Decreased perivascular fibrosis but not cardiac hypertrophy in ROCK1+/− haploinsufficient mice. Circulation 2005; 112: 2959–2965.
Masumoto A, Hirooka Y, Shimokawa H, Hironaga K, Setoguchi S, Takeshita A . Possible involvement of Rho-kinase in the pathogenesis of hypertension in humans. Hypertension 2001; 38: 1307–1310.
Masumoto A, Mohri M, Shimokawa H, Urakami L, Usui M, Takeshita A . Suppression of coronary artery spasm by the Rho-kinase inhibitor fasudil in patients with vasospastic angina. Circulation 2002; 105: 1545–1547.
Soga J, Hata T, Hidaka T, Fujii Y, Idei N, Fujimura N, Mikami S, Maruhashi T, Kihara Y, Chayama K, Kato H, Noma K, Liao JK, Higashi Y, . For ROCK investigator group. Rho-associated kinase (ROCK) activity, endothelial function and cardiovascular risk factors. Arterioscler Thromb Vasc Biol 2011; 31: 2353–2359.
Takemoto M, Sun J, Hiroki J, Shimokawa H, Liao JK . Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation 2002; 106: 57–62.
Ming XF, Viswambharan H, Barandier C, Ruffieux J, Kaibuchi K, Rusconi S, Yang Z . Rho GTPase/Rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Mol Cell Biol 2002; 22: 8467–8477.
Shimokawa H, Takeshita A . Rho-kinase is an important therapeutic target in cardiovascular medicine. Arterioscler Thromb Vasc Biol 2005; 25: 1767–1775.
Bailey SR, Mitra S, Flavahan S, Flavahan NA . Reactive oxygen species from smooth muscle mitochondria initiate cold-induced constriction of cutaneous arteries. Am J Physiol Heart Circ Physiol 2005; 289: H243–H250.
Jin L, Ying Z, Webb RC . Activation of Rho/Rho kinase signaling pathway by reactive oxygen species in rat aorta. Am J Physiol Heart Circ Physiol 2004; 287: H1495–H1500.
Rikitake Y, Liao JK . Rho-kinase mediates hyperglycemia-induced plasminogen activator inhibitor-1 expression in vascular endothelial cells. Circulation 2005; 111: 3261–3268.
Noma K, Goto C, Nishioka K, Jitsuiki D, Umemura T, Ueda K, Kimura M, Nakagawa K, Oshima T, Chayama K, Yoshizumi M, Liao JK, Higashi Y . Roles of ROCK and oxidative stress in the pathogenesis of aortic stiffness. J Am Coll Cardiol 2007; 49: 698–705.
Hernández-Perera O, Pérez-Sala D, Soria E, Lamas S . Involvement of Rho GTPases in the transcriptional inhibition of preproendothelin-1 gene expression by simvastatin in vascular endothelial cells. Circ Res 2000; 87: 616–622.
Sauzeau V, Le Jeune H, Cario-Toumaniantz C, Smolenski A, Lohmann SM, Bertoglio J, Chardin P, Pacaud P, Loirand G . Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J Biol Chem 2000; 275: 21722–21729.
Chitaley K, Webb RC . Nitric oxide induces dilation of rat aorta via inhibition of rho-kinase signaling. Hypertension 2002; 39: 438–442.
Hidaka T, Hata T, Soga J, Nakamura S, Fujii Y, Idei N, Fujimura N, Nishioka K, Kihara Y, Chayama K, Noma K, Liao JK, Higashi Y . Increased leukocyte Rho kinase (ROCK) and endothelial dysfunction in cigarette smokers. Hypertens Res 2010; 33: 354–359.
Fukata Y, Amano M, Kaibuchi K . Rho-Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol Sci 2001; 22: 32–39.
Noma K, Oyama N, Liao JK . Physiological role of ROCKs in the cardiovascular system. Am J Physiol Cell Physiol 2006; 290: C661–C668.
Satoh K, Fukumoto Y, Shimokawa H . Rho-kinase: important new therapeutic target in cardiovascular diseases. Am J Physiol Heart Circ Physiol 2011; 301: H287–H296.
Henderson E, Hardin CC, Walk SK, Tinoco I, Blackburn EH, Telomeric DNA . Oligonucleotides form novel intramolecular structures containing guanine-guanine base pairs. Cell 1987; 51: 899–908.
Blasco MA . Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet 2005; 6: 611–622.
Greider CW, Blackburn EH . The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell 1987; 51: 887–898.
Cohen SB, Graham ME, Lovrecz GO, Bache N, Robinson PJ, Reddel RR . Protein composition of catalytically active human telomerase from immortal cells. Science 2007; 315: 1850–1853.
Chang E, Harley CB . Telomere length and replicative aging in human vascular tissues. Proc Natl Acad Sci USA 1995; 92: 11190–11194.
Furumoto K, Inoue E, Nagao N, Hiyama E, Miwa N . Age-dependent telomere shortening is slowed down by enrichment of intracellular vitamin C via suppression of oxidative stress. Life Sci 1998; 63: 935–948.
Fuster JJ, Díez J, Andrés V . Telomere dysfunction in hypertension. J Hypertens 2007; 25: 2185–2192.
Zhu H, Belcher M, van der Harst P . Healthy aging and disease: role for telomere biology? Clin Sci 2011; 120: 427–440.
Okuda K, Khan MY, Skurnick J, Kimura M, Aviv H, Aviv A . Telomere attrition of the human abdominal aorta: relationships with age and atherosclerosis. Atherosclerosis 2000; 152: 391–398.
Sato I, Morita I, Kaji K, Ikeda M, Nagao M, Murota S . Reduction of nitric oxide producing activity associated with in vitro aging in cultured human umbilical vein endothelial cell. Biochem Biophys Res Commun 1993; 195: 1070–1076.
Matsushita H, Chang E, Glassford AJ, Cooke JP, Chiu CP, Tsao PS . eNOS activity is reduced in senescent human endothelial cells: Preservation by hTERT immortalization. Circ Res 2001; 89: 793–798.
Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I . Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation 2002; 105: 1541–1544.
Vasa M, Breitschopf K, Zeiher AM, Dimmeler S . Nitric oxide activates telomerase and delays endothelial cell senescence. Circ Res 2000; 87: 540–542.
Nakashima H, Ozono R, Suyama C, Sueda T, Kambe M, Oshima T . Telomere attrition in white blood cell correlating with cardiovascular damage. Hypertens Res 2004; 27: 319–325.
Haendeler J, Hoffmann J, Brandes RP, Zeiher AM, Dimmeler S . Hydrogen peroxide triggers nuclear export of telomerase reverse transcriptase via Src kinase family-dependent phosphorylation of tyrosine 707. Mol Cell Biol 2003; 23: 598–4610.
Brandes RP, Fleming I, Busse R . Endothelial aging. Cardiovasc Res 2005; 66: 286–294.
Serrano AL, Andrés V . Telomeres and cardiovascular disease: does size matter? Circ Res 2004; 94: 575–584.
Smogorzewska A, de Lange T . Different telomere damage signaling pathways in human and mouse cells. EMBO J 2002; 21: 4338–4348.
Minamino T, Komuro I . Vascular cell senescence: contribution to atherosclerosis. Circ Res 2007; 100: 15–26.
Scalera F, Borlak J, Beckmann B, Martens-Lobenhoffer J, Thum T, Täger M, Bode-Böger SM . Endogenous nitric oxide synthesis inhibitor asymmetric dimethyl L-arginine accelerates endothelial cell senescence. Arterioscler Thromb Vasc Biol 2004; 24: 1816–1822.
Acknowledgements
This study was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science and Culture of Japan, Japan Heart Foundation Grant for Research on Hypertension and Metabolism, and a Grant for Research Foundation for Community Medicine.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Higashi, Y., Kihara, Y. & Noma, K. Endothelial dysfunction and hypertension in aging. Hypertens Res 35, 1039–1047 (2012). https://doi.org/10.1038/hr.2012.138
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2012.138
Keywords
This article is cited by
-
Germline predictors for bevacizumab induced hypertensive crisis in ECOG-ACRIN 5103 and BEATRICE
British Journal of Cancer (2024)
-
Pathophysiology and probable etiology of cerebral small vessel disease in vascular dementia and Alzheimer’s disease
Molecular Neurodegeneration (2023)
-
Strength training for arterial hypertension treatment: a systematic review and meta-analysis of randomized clinical trials
Scientific Reports (2023)
-
Vascular function: a key player in hypertension
Hypertension Research (2023)
-
Current Management of Hypertension in Older Adults
Drugs & Aging (2023)
