
Abstract
Pirfenidone (5-methyl-1-phenyl-2-[1H]-pyridone) is an effective drug for idiopathic interstitial pneumonia that can prevent and reverse tissue fibrosis in several organs. Therefore, we investigated whether pirfenidone has a potential role in preventing angiotensin II (Ang II)-induced cardiac hypertrophy. A cardiac hypertrophic mouse model was created using an Ang II infusion (200 ng kg−1 min−1) in wild-type mice for 2 weeks. Mice were divided into the following three groups: a saline-infused (control) group, an Ang II infusion (vehicle) group and an Ang II infusion+pirfenidone-treated (PFD) group, which received pirfenidone (300 mg kg−1 per day) by gastric gavage during the Ang II infusion. At 2 weeks, we assessed hemodynamics and cardiac function and investigated tissue fibrosis of the myocardium histologically and genetically. Blood pressure in the vehicle group was significantly increased compared to the control group. Although blood pressure was not different between the vehicle and PFD groups, heart weight was significantly decreased in the PFD group. Echocardiography revealed that left ventricular hypertrophy was significantly increased in the vehicle group vs. the control group. Interestingly, pirfenidone significantly inhibited this effect. Continuous infusion of Ang II increased the perivascular and interstitial tissue fibrosis, and pirfenidone inhibited these fibrotic changes. Pirfenidone also inhibited Ang II-induced hypertrophy. In the vehicle group, the mRNA expressions of atrial natriuretic peptide, brain natriuretic peptide and transforming growth factor-β1 were increased, which was significantly inhibited by pirfenidone. Furthermore, the expression of mineralocorticoid receptors was attenuated by pirfenidone. These results indicate that pirfenidone might be effective as an antifibrotic drug in the treatment of cardiac hypertrophy induced by hypertension.
Similar content being viewed by others

Introduction
Cardiovascular damage due to hypertension represents a growing public health problem. Vascular remodeling is an adaptive response to increased arterial blood pressure. Angiotensin II (Ang II), an important effector of the renin–angiotensin–aldosterone system, is one of the most important factors involved in arterial remodeling.1 In addition to its role in arterial blood pressure regulation, Ang II induces vascular smooth muscle cell hyperplasia and hypertrophy through the transactivation of epidermal growth factor, platelet-derived growth factor and insulin-like growth factor I receptors.2 Ang II is also critically involved in the initial stage of inflammation, increased vascular permeability and leukocyte recruitment through the expression of vascular endothelial growth factor and adhesion molecules or chemokines, respectively.1, 3 Ang II also plays a critical role in the genesis of cardiac hypertrophy and fibrosis by producing transforming growth factor (TGF)-β, platelet-derived growth factor and endothelin-1, as well as by attenuating interstitial matrix metalloproteinase activity, resulting in left ventricular (LV) dysfunction. Most of these effects are mediated by the type 1 Ang II receptor (AT1R) rather than the type 2 Ang II receptor. The Ang II/AT1 receptor interaction generates an immediate calcium-dependent response and late nuclear factor-κB activation through the activation of different pathways, including protein kinase C, mitogen-activated protein kinase cascades, receptor tyrosine kinases and non-receptor tyrosine kinases.2, 4
Pirfenidone (5-methyl-1-phenyl-2-[1H]-pyridone) has been shown to reduce the fibrosis of different organs (for example, lung5, 6 kidney7, 8, 9 liver,10 heart11, 12, 13) and vascular remodeling14 through its inhibitory effect on fibroblast growth and collagen synthesis, which is achieved by reducing the expression of profibrotic cytokines (for example, TGF-β15, 16). Furthermore, pirfenidone has been successfully used in a double-blind, placebo-controlled trial focused on patients with idiopathic pulmonary fibrosis.17 However, the role of pirfenidone on hypertensive heart diseases is still unknown.
In this study, we investigated the effect of pirfenidone on Ang II-induced ventricular dysfunctions, including hypertrophy, fibrosis and coronary arterial remodeling in vivo.
Methods
Animals and experimental design
All procedures involving animals were approved by the animal care and use committee of Osaka City University. Experiments were performed with 8- to 9- week-old male BALB/c mice (CLEA Japan, Tokyo, Japan). Pirfenidone was provided as a gift from Shionogi (Tokyo, Japan).
To induce cardiac hypertrophy, Ang II (200 ng kg−1 min−1) was dissolved in saline and continuously infused subcutaneously over 14 days via an osmotic mini-pump (ALZET, Cupertino, CA, USA). The control group received a saline infusion. Ang II-infused mice were divided into two groups. Pirfenidone (300 mg kg−1 per day, the PFD group) or a 0.5% carboxymethylcellulose solution (the vehicle group) was administered orally by gastric gavage once a day for 14 days (started on the same day as the Ang II infusion). The systolic blood pressure of conscious mice was measured using the tail-cuff method (BP98A; Softron, Tokyo, Japan) before and 13 days after the Ang II infusion.
On day 14 after the infusion, cardiac function was examined using echocardiography as described below. The hearts were immediately excised afterwards, and the ventricle was separated from the atrium and weighed. The ventricle was divided into upper and lower portions. Subsequently, the upper portion of the LV was immediately frozen in liquid nitrogen and stored at −80°C until use. The lower portion was set overnight in 4% paraformaldehyde and was embedded in paraffin.
Echocardiographic study
Echocardiographic studies were performed on mice using the SONOS 5500 (Philips, Best, the Netherlands) as described previously.18, 19 In brief, mice were lightly anesthetized with tiletamine (10 mg kg−1 intraperitoneally) and xylazine (10 mg kg−1 intraperitoneally). A two-dimensional, short-axis view of the LV was obtained at the level of the papillary muscles. Pulsed wave Doppler spectra (E and A waves) of mitral inflow velocities were recorded from the apical four-chamber view with the sample volume placed near the tips of the mitral leaflets and adjusted to the position of maximum velocity and laminar flow pattern. All Doppler spectra were recorded at a paper speed of 200 mm s−1 and analyzed offline.
Estimation of cardiac fibrosis
The area of perivascular and interstitial fibrosis was measured as described previously.19, 20 In short, the mouse heart was set with 10% formaldehyde overnight and embedded in paraffin. Five-μm-thick sections were cut and stained with Sirius red stain to measure the interstitial fibrosis area. The interstitial fibrosis area was calculated as the ratio of the sum of the total area of interstitial fibrosis to the sum of the total connective tissue area plus the cardiomyocyte area in all the LV fields of the section that excluded perivascular fibrosis. Perivascular fibrosis was assessed by calculating the ratio of the area of collagen-stained material to the total vessel area, which was defined as the medial plus luminal area. Each field was analyzed with the image-analyzing software (Micro Analyzer, Nihon Poladigital, Tokyo, Japan).
RNA preparation and analysis
RNA from the LV was isolated using ISOGEN (Nippon Gene, Toyama, Japan).18 To elucidate the gene expression levels, we subjected the RNA samples to quantitative real-time polymerase chain reaction (ABI Prism 7700; Perkin-Elmer Applied Biosystems, Carlsbad, CA, USA). One-step quantitative real-time polymerase chain reaction reactions were performed using 100 ng of RNA per reaction. TaqMan primers and probes were designed using Primer 3 (v. 0.4.0); see Table 1. For analysis, transcript levels were normalized to glyceraldehyde-3-phosphate dehydrogenase.
Statistical analysis
All data are presented as means±s.e.m. Comparisons among groups were made using a one-way analysis of variance followed by Fisher's protected least significant difference test using StatView (SAS Institute, Cary, NC, USA). A Pearson's correlation coefficient was used for correlation analysis. Differences were considered to be statistically significant at a value of P<0.05.
Results
Effect of pirfenidone on blood pressure, body weight and heart weight
The blood pressure and body weight of the mice is shown in Figures 1a and b. The systolic blood pressure of the mice receiving a saline infusion (the control group), Ang II infusion (the vehicle group) and Ang II infusion+PFD treatment (the PFD group) were 110±2, 167±6 and 167±19 mm Hg, respectively, at day 13. Thus, blood pressure elevation by Ang II was not inhibited by pirfenidone. Body weight was not affected by pirfenidone, although significantly decreased by Ang II.

Systolic blood pressure (a) and body weights (b) of mice before and after 2 weeks of Ang II administration (200 ng kg−1 min−1) and pirfenidone (PFD) treatment. Co indicates saline-infused control mice; Ve indicates Ang II-infused mice treated with vehicle; and PFD indicates Ang II-infused mice treated with PFD. Values represent means±s.e.m. (n=6–10).*P<0.05 vs. control mice at each time point. Representative images of light micrographs of cross-sections of the mid-portion of the heart (c) (bar=1 mm) and heart weight (d) from saline-infused control group (Co), and Ang II-infused groups treated with Ve or PFD. Values represent means±s.e.m. (n=6–10). *P<0.05 vs. Co; #P<0.05 when Ve vs. PFD.
Effect of pirfenidone on cardiac hypertrophy in Ang II-infused mice
The effect of pirfenidone on Ang II-induced LV hypertrophy is shown in Figures 1c and d. Ang II infusion for 14 days significantly increased the heart weight compared to the control (0.100±0.002 vs. 0.120±0.006 g, respectively, P<0.01), and the effect of Ang II was significantly inhibited by pirfenidone (0.108±0.002 g). Intraventricular septum and posterior wall thickness estimated by echocardiography were also significantly larger in the vehicle and the PFD groups than in the control group (Table 2). The increase in thickness was significantly smaller in the PFD group than the vehicle group. LV end-diastolic and -systolic dimension, fractional shortening and ejection fraction were not significantly different among the groups.
Effects of pirfenidone on perivascular fibrosis in Ang II-infused mice
Perivascular fibrosis of large and small coronary arteries in the LV of mice was larger than that of the control mice (Figures 2a and d). Perivascular fibrosis caused by Ang II was attenuated by pirfenidone treatment.

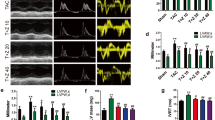
Perivascular fibrosis and myocardial interstitial fibrosis in control, Ang II-infused and pirfenidone (PFD)-treated mice. Representative images of light micrographs of cross-sections of the coronary artery (a) and myocardial interstitial fibrosis (b) on left ventricular free wall sections. Original magnification: a, × 400 (bar=50 μm) and b, × 200. Co indicates saline-infused control mice; Ve indicates Ang II-infused mice treated with vehicle; and PFD indicates Ang II-infused mice treated with PFD. (c) Representative pulse-wave Doppler spectra of mitral inflow pattern from the saline-infused control group (Co), and Ang II-infused groups treated with Ve or PFD. Quantitative results of perivascular fibrosis (d) and relative area of interstitial fibrosis (e). Co indicates saline-infused control mice; Ve indicates Ang II-infused mice treated with vehicle; and PFD indicates Ang II-infused mice treated with PFD. Values represent means±s.e.m. (n=6–10). *P<0.05 vs. Co; #P<0.05 when Ve vs. PFD.
Effects of pirfenidone on myocardial interstitial fibrosis and LV diastolic function in Ang II-infused mice
Ang II infusion for 2 weeks significantly increased the area of myocardial interstitial fibrosis in mice by 1.8-fold (P<0.01), whereas Ang II-induced fibrosis was significantly diminished by pirfenidone (Figures 2b and e).
The mitral inflow pattern in each group, as estimated by echocardiography, is shown in Figure 2c and Table 2. The ratio of the early rapid filling wave (E wave) velocity to the atrial contraction (A wave) velocity (E/A ratio) in the vehicle group was expressed as an abnormal pattern. Ang II lessened the E wave velocity and E/A ratio. Pirfenidone tended to improve them.
Effect of pirfenidone on cardiac gene expressions
The LV mRNA levels measured by quantitative real-time polymerase chain reaction in each group of mice are shown in Figure 3. LV mRNA expression levels for atrial natriuretic peptide and brain natriuretic peptide, which are closely related to cardiac hypertrophy, were increased by 9.1- and 3.0-fold (P<0.01), respectively, following Ang II infusion. The upregulation of these mRNAs by Ang II was significantly decreased by pirfenidone. The mRNA levels for LV TGF-β1 and monocyte chemoattractant protein-1 (MCP-1), which are closely related to cardiac fibrosis and remodeling, were increased by 1.8- and 2.2-fold (P<0.01), respectively, following Ang II infusion. Pirfenidone significantly attenuated the increased TGF-β1 expression. The MCP-1 expression level, which was increased by Ang II, tended to be suppressed with the treatment of pirfenidone. The expression level of interferon-γ, which has several potential antifibrotic actions, was significantly reduced by Ang II and increased with pirfenidone treatment. Peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α), which is closely related to mitochondrial function, was also downregulated by Ang II, and pirfenidone tended to reverse this effect.

Gene expressions in control, Ang II-infused and pirfenidone (PFD)-treated mice. The bar graph shows each mRNA value, corrected for the glyceraldehyde-3-phosphate dehydrogenase mRNA value. Mean values in the control mice group are represented as 1. Co indicates saline-infused control mice; Ve indicates Ang II-infused mice treated with vehicle; and PFD indicates Ang II-infused mice treated with PFD. Values represent means±s.e.m. (n=6–10). *P<0.05 vs. Co; #P<0.05 when Ve vs. PFD.
In addition, we investigated the role of pirfenidone on the gene expressions of Ang II type 1a receptor (AT1aR) and mineralocorticoid receptor (MR) in the mouse heart. Ang II significantly reduced the expression levels of AT1aR and MR. Pirfenidone did not affect the AT1aR expression. However, the MR expression in the PFD group was further decreased compared to that in the vehicle group. The expression of type 1 Ang II receptor in each group was too low to compare (data not shown).
Correlation of effects of cardiac TGF-β1 expression and perivascular fibrosis or myocardial interstitial fibrosis
We analyzed the correlation of the effects of cardiac TGF-β1 expression and LV hypertrophy, perivascular fibrosis or myocardial interstitial fibrosis. A correlation between TGF-β1 expression level and heart weight was not found (data not shown). However, there was a positive correlation between perivascular or myocardial interstitial fibrosis and TGF-β1 expression level (Figure 4). These data suggest that the expression level of TGF-β1 parallels the perivascular and myocardial interstitial fibrotic changes.

The correlation between the TGF-β1 expression level and the ratio of perivascular fibrosis (left and middle panels) or myocardial interstitial fibrosis (right panel).
Discussion
Accumulating evidence suggests that TGF-β plays a causal role in LV hypertrophy induced by pressure overload, which leads to LV systolic and diastolic dysfunction.21 Cardiac hypertrophy results in an increased expression of the mRNA for the extracellular matrix protein collagen.22 Eghbali et al.23 have demonstrated that TGF-β1 regulates collagen biosynthesis, and, subsequently, the actual deposition of collagen proteins in cultured cardiac fibroblasts. Furthermore, Villarreal et al.24 have reported that the mRNA level of TGF-β1 was increased in cardiac hypertrophy induced by thoracic aorta banding in rats. These results suggest that TGF-β1 is one of the major regulators of extracellular matrix protein deposition. Another study has reported that TGF-β1 appears to play an important role directly or in part via increased β-adrenergic signaling during the transition from compensated to decompensated cardiac hypertrophy.25 Thus, blocking TGF-β1 pathways might be a pharmacological intervention in cardiac remodeling involving cardiac hypertrophy.
Pirfenidone has been reported to downregulate the gene expression and protein production of TGF-β,6 although its mechanisms of action are not well understood. The antifibrotic actions of pirfenidone have been shown in several organs, including bleomycin-induced fibrotic lung in hamsters,26 remnant kidney with collagen accumulation by partial nephrectomy in rats,27 dimethylnitrosamine-induced fibrotic liver in rats,28 chlorhexidine gluconate and ethanol-induced sclerosing peritonitis in rats29 and intimal thickening after angioplasty of the carotid artery in rats.14
There are a few reports concerning the effect of pirfenidone on heart diseases. Lee et al.30 investigated the effects of pirfenidone on arrhythmogenic atrial remodeling with ventricular tachypacing-induced chronic heart failure in canines. In their study, pirfenidone treatment resulted in a significant reduction of atrial fibrosis. Mirkovic et al.11 reported that pirfenidone reverses and prevents cardiac remodeling as well as increased cardiac stiffness in deoxycorticosterone acetate-salt hypertensive rats. Those results suggest a broad effect of pirfenidone on potent profibrotic mediators.31 However, there has been no evidence regarding the role of pirfenidone in cardiac hypertrophy and remodeling or its genetic analysis in vivo. In this study, we determined the role of pirfenidone in hypertrophy and remodeling that is induced by Ang II infusion. Mice were infused with Ang II, which caused a gradual elevation of blood pressure and gradual development of cardiac hypertrophy; this finding is similar to the physiological condition in humans.
In this study, we first showed that pirfenidone significantly inhibited Ang II-induced LV TGF-β1 expression and LV hypertrophy (Figures 1 and 3). Furthermore, there was a positive correlation between perivascular or myocardial interstitial fibrosis and TGF-β1 expression level. These results suggest that LV TGF-β1 is directly suppressed by pirfenidone (Figure 4). TGF-β signaling is thought to be a therapeutic target for fibrotic diseases.32 Iyer et al.6 suggested that pirfenidone may directly act on both macrophages and epithelial cells and fibroblasts to compromise their ability to synthesize and release TGF-β1. However, we could not testify the role of these cells because this study was based on whole-heart analysis in vivo. Further studies are needed to elucidate this point.
As estimated by LV weight, perivascular and interstitial fibrosis, echocardiographic measurements, and LV atrial natriuretic peptide and brain natriuretic peptide mRNA expression levels, cardiac hypertrophy induced by Ang II was significantly attenuated in pirfenidone-treated mice independently of blood pressure. These data strongly support the hypothesis that pirfenidone could improve the progression of cardiac hypertrophy and remodeling induced by Ang II. Furthermore, Doppler echocardiographic analysis of the LV inflow pattern revealed that Ang II induced an ‘abnormal relaxation pattern’, and pirfenidone returned it to a ‘normal pattern’ (Figure 2c); however, it was difficult to evaluate the LV inflow pattern quantitatively because the heart rates were not equalized adequately in the echocardiographic study. Therefore, pirfenidone treatment effectively prevents an increase in LV relaxation and a decrease in the passive elastic properties.
In addition to Ang II-induced LV TGF-β1 expression, we also examined some LV mRNA expression levels. MCP-1 is one of the major inflammatory mediators. Pressure overload-induced perivascular and cardiac fibrosis are mediated through MCP-1 induction and macrophage accumulation.33 In addition, MCP-1 expression is increased in the aortic tissues of hypertensive rats34 and in human atherosclerotic lesions.35 Thus, MCP-1 plays an important role in the process of cardiac hypertrophy and fibrosis. In this study, LV MCP-1 expression was significantly increased by Ang II in accordance with our previous report,36 and the increase tended to be smaller in the LV of pirfenidone-treated mice. It was not obvious whether pirfenidone could inhibit cardiac MCP-1 expression in this study.
The expression of PGC-1α, which is a critical transcriptional regulator of energy metabolism, is decreased with the development of cardiac dysfunction and heart failure. Accordingly, PGC-1α is thought to be a novel therapeutic target for heart failure.37 In our model, Ang II decreased the level of PGC-1α expression, and pirfenidone reversed this effect. Thus, pirfenidone may inhibit the development of cardiac dysfunction partially via PGC-1α.
It is well-known that Ang II induces cardiac remodeling via not only type 1 Ang II receptor but also MR.38 Therefore, we investigated whether pirfenidone affected gene expressions of AT1aR and MR in the mouse heart (Figure 3). Excessive administration of Ang II reduced the expressions of both AT1aR and MR. Pirfenidone decreased the MR expression more, suggesting that it could prevent Ang II-induced cardiac remodeling partially via the inhibition of aldosterone signaling pathways.
In conclusion, our present work provided in vivo evidence that the antifibrotic agent pirfenidone prevented the progression of Ang II-induced cardiac hypertrophy and fibrosis. One of the major mechanisms of pirfenidone may be mediated via TGF-β1 expression and, partially, via MR expression.
References
Duprez DA . Role of the renin–angiotensin–aldosterone system in vascular remodeling and inflammation: a clinical review. J Hypertens 2006; 24: 983–991.
Mehta PK, Griendling KK . Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 2007; 292: C82–C97.
Touyz RM . Molecular and cellular mechanisms in vascular injury in hypertension: role of angiotensin II. Curr Opin Nephrol Hypertens 2005; 14: 125–131.
Kim S, Iwao H . Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol Rev 2000; 52: 11–34.
Azuma A . Pirfenidone: antifibrotic agent for idiopathic pulmonary fibrosis. Expert Rev Respir Med 2010; 4: 301–310.
Iyer SN, Gurujeyalakshmi G, Giri SN . Effects of pirfenidone on transforming growth factor-beta gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J Pharmacol Exp Ther 1999; 291: 367–373.
Cho ME, Kopp JB . Pirfenidone: an anti-fibrotic therapy for progressive kidney disease. Expert Opin Invest Drugs 2010; 19: 275–283.
Decleves AE, Sharma K . New pharmacological treatments for improving renal outcomes in diabetes. Nat Rev Nephrol 2010; 6: 371–380.
RamachandraRao SP, Zhu Y, Ravasi T, McGowan TA, Toh I, Dunn SR, Okada S, Shaw MA, Sharma K . Pirfenidone is renoprotective in diabetic kidney disease. J Am Soc Nephrol 2009; 20: 1765–1775.
Grizzi F . Pirfenidone: a potential therapeutic option in the treatment of liver fibrosis. Clin Exp Pharmacol Physiol 2009; 36: 961–962.
Mirkovic S, Seymour AM, Fenning A, Strachan A, Margolin SB, Taylor SM, Brown L . Attenuation of cardiac fibrosis by pirfenidone and amiloride in DOCA-salt hypertensive rats. Br J Pharmacol 2002; 135: 961–968.
Van Erp C, Irwin NG, Hoey AJ . Long-term administration of pirfenidone improves cardiac function in mdx mice. Muscle Nerve 2006; 34: 327–334.
Nguyen DT, Ding C, Wilson E, Marcus GM, Olgin JE . Pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias. Heart Rhythm 2010; 7: 1438–1445.
Backes A, Seay U, Sedding DG, Tillmanns HH, Braun-Dullaeus RC . Inhibition of matrix deposition: a new strategy for prevention of restenosis after balloon angioplasty. J Cardiovasc Pharmacol 2010; 55: 213–218.
Burghardt I, Tritschler F, Opitz CA, Frank B, Weller M, Wick W . Pirfenidone inhibits TGF-beta expression in malignant glioma cells. Biochem Biophys Res Commun 2007; 354: 542–547.
Dosanjh A . Pirfenidone: anti-fibrotic agent with a potential therapeutic role in the management of transplantation patients. Eur J Pharmacol 2006; 536: 219–222.
Azuma A, Nukiwa T, Tsuboi E, Suga M, Abe S, Nakata K, Taguchi Y, Nagai S, Itoh H, Ohi M, Sato A, Kudoh S . Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2005; 171: 1040–1047.
Izumi Y, Okatani H, Shiota M, Nakao T, Ise R, Kito G, Miura K, Iwao H . Effects of metoprolol on epinephrine-induced takotsubo-like left ventricular dysfunction in non-human primates. Hypertens Res 2009; 32: 339–346.
Izumiya Y, Kim S, Izumi Y, Yoshida K, Yoshiyama M, Matsuzawa A, Ichijo H, Iwao H . Apoptosis signal-regulating kinase 1 plays a pivotal role in angiotensin II-induced cardiac hypertrophy and remodeling. Circ Res 2003; 93: 874–883.
Kim-Mitsuyama S, Izumi Y, Izumiya Y, Yoshida K, Yoshiyama M, Iwao H . Additive beneficial effects of the combination of a calcium channel blocker and an angiotensin blocker on a hypertensive rat-heart failure model. Hypertens Res 2004; 27: 771–779.
Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, Imaizumi T . Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation 2002; 106: 130–135.
Chapman D, Weber KT, Eghbali M . Regulation of fibrillar collagen types I and III and basement membrane type IV collagen gene expression in pressure overloaded rat myocardium. Circ Res 1990; 67: 787–794.
Eghbali M, Tomek R, Sukhatme VP, Woods C, Bhambi B . Differential effects of transforming growth factor-beta 1 and phorbol myristate acetate on cardiac fibroblasts. Regulation of fibrillar collagen mRNAs and expression of early transcription factors. Circ Res 1991; 69: 483–490.
Villarreal FJ, Dillmann WH . Cardiac hypertrophy-induced changes in mRNA levels for TGF-beta 1, fibronectin, and collagen. Am J Physiol 1992; 262: H1861–H1866.
Rosenkranz S, Flesch M, Amann K, Haeuseler C, Kilter H, Seeland U, Schluter KD, Bohm M . Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-beta(1). Am J Physiol Heart Circ Physiol 2002; 283: H1253–H1262.
Iyer SN, Wild JS, Schiedt MJ, Hyde DM, Margolin SB, Giri SN . Dietary intake of pirfenidone ameliorates bleomycin-induced lung fibrosis in hamsters. J Lab Clin Med 1995; 125: 779–785.
Shimizu T, Fukagawa M, Kuroda T, Hata S, Iwasaki Y, Nemoto M, Shirai K, Yamauchi S, Margolin SB, Shimizu F, Kurokawa K . Pirfenidone prevents collagen accumulation in the remnant kidney in rats with partial nephrectomy. Kidney Int Suppl 1997; 63: S239–S243.
Tada S, Nakamuta M, Enjoji M, Sugimoto R, Iwamoto H, Kato M, Nakashima Y, Nawata H . Pirfenidone inhibits dimethylnitrosamine-induced hepatic fibrosis in rats. Clin Exp Pharmacol Physiol 2001; 28: 522–527.
Suga H, Teraoka S, Ota K, Komemushi S, Furutani S, Yamauchi S, Margolin S . Preventive effect of pirfenidone against experimental sclerosing peritonitis in rats. Exp Toxicol Pathol 1995; 47: 287–291.
Lee KW, Everett THt, Rahmutula D, Guerra JM, Wilson E, Ding C, Olgin JE . Pirfenidone prevents the development of a vulnerable substrate for atrial fibrillation in a canine model of heart failure. Circulation 2006; 114: 1703–1712.
Sata Y, Krum H . The future of pharmacological therapy for heart failure. Circ J 2010; 74: 809–817.
Ruiz-Ortega M, Rodriguez-Vita J, Sanchez-Lopez E, Carvajal G, Egido J . TGF-beta signaling in vascular fibrosis. Cardiovasc Res 2007; 74: 196–206.
Kai H, Mori T, Tokuda K, Takayama N, Tahara N, Takemiya K, Kudo H, Sugi Y, Fukui D, Yasukawa H, Kuwahara F, Imaizumi T . Pressure overload-induced transient oxidative stress mediates perivascular inflammation and cardiac fibrosis through angiotensin II. Hypertens Res 2006; 29: 711–718.
Capers Qt, Alexander RW, Lou P, De Leon H, Wilcox JN, Ishizaka N, Howard AB, Taylor WR . Monocyte chemoattractant protein-1 expression in aortic tissues of hypertensive rats. Hypertension 1997; 30: 1397–1402.
Reape TJ, Groot PH . Chemokines and atherosclerosis. Atherosclerosis 1999; 147: 213–225.
Kim-Mitsuyama S, Izumi Y, Izumiya Y, Namba M, Yoshida K, Wake R, Yoshiyama M, Iwao H . Dominant-negative c-Jun inhibits rat cardiac hypertrophy induced by angiotensin II and hypertension. Gene Therapy 2006; 13: 348–355.
Sano M, Izumi Y, Helenius K, Asakura M, Rossi DJ, Xie M, Taffet G, Hu L, Pautler RG, Wilson CR, Boudina S, Abel ED, Taegtmeyer H, Scaglia F, Graham BH, Kralli A, Shimizu N, Tanaka H, Makela TP, Schneider MD . Menage-a-trois 1 is critical for the transcriptional function of PPARgamma coactivator 1. Cell Metab 2007; 5: 129–142.
Lemarie CA, Paradis P, Schiffrin EL . New insights on signaling cascades induced by cross-talk between angiotensin II and aldosterone. J Mol Med 2008; 86: 673–678.
Acknowledgements
We thank Chiori Asahi for technical assistance. This study was partially supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (YI).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Yamazaki, T., Yamashita, N., Izumi, Y. et al. The antifibrotic agent pirfenidone inhibits angiotensin II-induced cardiac hypertrophy in mice. Hypertens Res 35, 34–40 (2012). https://doi.org/10.1038/hr.2011.139
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2011.139
Keywords
This article is cited by
-
Human amnion epithelial cell therapy reduces hypertension-induced vascular stiffening and cognitive impairment
Scientific Reports (2024)
-
Pirfenidone ameliorates liver steatosis by targeting the STAT3-SCD1 axis
Inflammation Research (2023)
-
Pirfenidone as a novel cardiac protective treatment
Heart Failure Reviews (2022)
-
Pirfenidone in heart failure with preserved ejection fraction: a randomized phase 2 trial
Nature Medicine (2021)
-
Myocardial Fibrosis in Heart Failure: Anti-Fibrotic Therapies and the Role of Cardiovascular Magnetic Resonance in Drug Trials
Cardiology and Therapy (2020)
