
Abstract
The relationship between obesity and hypertension is well established both in children and adults. The mechanisms through which obesity directly causes hypertension are still an area of research. Activation of the sympathetic nervous system has been considered to have an important function in the pathogenesis of obesity-related hypertension. The arterial-pressure control mechanism of diuresis and natriuresis, according to the principle of infinite feedback gain, seems to be shifted toward higher blood-pressure levels in obese individuals. During the early phases of obesity, primary sodium retention exists as a result of increase in renal tubular reabsorption. Extracellular-fluid volume is expanded and the kidney-fluid apparatus is resetted to a hypertensive level, consistent with a model of hypertension because of volume overload. Plasma renin activity, angiotensinogen, angiotensin II and aldosterone values display significant increase during obesity. Insulin resistance and inflammation may promote an altered profile of vascular function and consequently hypertension. Leptin and other neuropeptides are possible links between obesity and the development of hypertension. Obesity should be considered as a chronic medical condition, which is likely to require long-term treatment. Understanding of the mechanisms associated with obesity-related hypertension is essential for successful treatment strategies.
Similar content being viewed by others
Introduction
Obesity is a common disorder that develops from the interaction between the genotype and the environment and involves social, behavioral, cultural, physiological, metabolic and genetic factors. A large number of studies have shown that obesity has an important negative impact on health in a population, leading to the recommendation for general practitioners to have an important function in the management of this condition and of its associated comorbidities such as hypertension, hyperlipidemia and hyperinsulinemia/insulin resistance. The relationship between obesity and hypertension is well established both in adults and children.1, 2 Obese individuals exhibit higher levels of office as well as ambulatory blood pressure (BP) from childhood to old age. Obese subjects display higher BP levels than non-obese individuals even in the normotensive range. The combination of obesity, hypertension and other cardiovascular risk factors significantly increases the probability of adverse cardiovascular outcomes, and raises considerations for aggressive treatment strategies.3
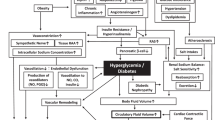
The mechanisms through which obesity directly causes hypertension are still an area of research. Human and animal studies have elucidated the function of adipose tissue derivatives (adipokines and cytokines), neurohumoral pathways, metabolic functions and modulation of pressor/depressor mechanisms. Although obesity-related hypertension may be the result of a combination or overlap of a number of these factors, a systematic review of the data concerning each of the possible mechanism would enhance our understanding of its contribution to the progression of the disease (Figure 1).

Mechanisms involved in the pathogenesis of obesity-induced hypertension. PAI-1, plasminogen activator inhibitor-1; Tx-A2, thromboxane A2; IL-6, interleukin-6; IL-1β, interleukin-1β; TNFα, tumor necrosis factor-α; CRP, C-reactive protein; ROS, reactive oxygen species; FFAs, free-fatty acids; VCAM-1, vascular cell adhesion molecule-1; ICAM-1, inter-cellular adhesion molecule-1; NO, nitric oxide; ET-1, endothelin-1; RAS, rennin–angiotensin system; SNS, sympathetic nervous system; AgRP, agouti-related peptide; NPY, neuropeptide Y; POMC, proopiomelanocortin; ARC, arcuate nucleus; α-MSH, α-melanocyte-stimulating hormone; MC3R, melanocortin 3 receptor; MC4R, melanocortin 4 receptor.
Sympathetic activation in obesity
Activation of the sympathetic nervous system (SNS), measured with direct or indirect methods, has been considered to have a crucial function in the pathogenesis of hypertension among obese individuals. Microneurograhic techniques, a direct method of measuring increased SNS activity, provide evidence of high muscle SNS activity in obese subjects.4 High-caloric intake increases norepinephrine turnover in peripheral tissues, raises resting plasma norepinephrine concentrations—an indirect measurement of SNS activity—and amplifies the rise of plasma norepineprhine in response to stimuli such as upright posture.5 High dietary content in fat and carbohydrate has been suggested to acutely stimulate peripheral a1- and b-adrenergic receptors, leading to elevated sympathetic activity and hypertension.6 A cafeteria diet has been reported to upregulate hypothalamic tyrosine hydroxylase and the hypothalamic adrenoceptor gene expression of the [α]2B receptor in obese hypertensive rats.7 Furthermore, pharmacological blockade of adrenergic activity in experimental models (combined a- and b-adrenergic blockade or central sympathetic modulation by clonidine) markedly blunted the rise of BP in dogs fed a high-fat diet.8 Similar results have been reported for human beings. Combined a- and b-adrenergic blockade significantly reduced BP in obese compared with lean patients with essential hypertension.9 However, elevated sympathetic activity did not seem to account for the increased heart rate in obesity. Instead, the elevated heart rate seems to be the effect of decreased parasympathetic activity.10 Inversely, modest weight reduction is able to suppress the activity of the SNS. It has been suggested, however, that an elevated efferent sympathetic activity in the peroneal nerves may not characterize all obese patients, but only those concomitantly affected by obstructive sleep apnea, an issue, which is still a matter of debate.11, 12
There has been one reported exception in the findings of high SNS activity with increasing adiposity. Pima Indians have the highest reported prevalence of obesity and hyperinsulinemia in the world, but a relatively low presence of hypertension and atherosclerotic disease. The low muscle SNS activity that has been reported in Pima Indians indicates the possible differences between ethnic groups and confirms the physiological importance of the SNS upregulation as an important mechanism of obesity-related hypertension.13 The mechanisms that have been proposed to be responsible for an increased sympathetic activity in obesity include impaired function of the baroreceptor sensitivity, increased levels of circulating free-fatty acids (FFAs), angiotensin (Ang) II, insulin and leptin.
The arterial baroreceptors acutely respond to increases in BP by parasympathetic activation and sympathetic inhibition. A reduced sensitivity of the arterial baroreflex manifested as a diminished response in heart rate to BP changes may occur in long-standing hypertension and obesity,4 presumably because of various mechanisms, including a concomitant increase in central sympathetic outflow and the effects of arteriosclerotic lesions leading to an increased stiffness of the large arteries in which the receptors are located. We have earlier reported an increased carotid arteries intima media thickness in obese subjects independently of the BP levels, even in a normotensive BP range, suggesting an early course of the atherosclerotic process in obesity.14 Impaired baroreflex sensitivity leads to withdrawal of parasympathetic cardiac modulation, which occurs in obesity even in the absence of an elevation in arterial pressure.
Elevated levels of FFAs have been reported in obese hypertensive subjects. Abnormal distribution of FFAs in obese patients has been found to enhance vascular a-adrenergic sensitivity and consequently to increase a-adrenergic tone.15 The exact mechanisms through which endogenous FFAs exert this effect have not been thoroughly investigated. Lysophospholipids and FFAs inhibit Na+, K+-ATPase and the sodium pump raising vascular smooth muscle tone and resistance.16 Binding of lysophospholipids and FFAs to Na/K-ATPase changes the interaction of the enzyme with neighboring membrane proteins and induces the formation of multiple signaling modules. As a result, the epidermal growth factor receptor is activated and production of reactive oxygen species is increased. Another target system of the FFAs is the calcium-independent isoenzyme of protein kinase C, which is a vital element in mediating signal transduction and cell regulation. FFAs act as potent activators of phosphorylation of protein kinase C.17 Other studies have reported a direct action of FFAs released from phospholipids on ion channels at the cellular membranes of smooth muscle cells and other tissues.18
Hall et al.19 have reported that plasma renin activity displays a significant increase during obesity, despite extracellular-fluid volume expansion and sodium retention in dogs fed with a high-fat diet. Moreover, studies in patients under sodium restriction, which activates the rennin–Ang system (RAS), provided evidence for a presynaptic potentiating effect of Ang II on sympathetic neurotransmission.20
Renal mechanisms
Impairment of pressure natriuresis
The arterial pressure control mechanism of diuresis and natriuresis according to the principle of infinite feedback gain seems to be shifted toward higher BP values in obese patients.21 Abnormalities in these mechanisms that would tend to raise BP increase sodium and water excretion through pressure natriuresis and diuresis. As long as excretion exceeds intake, extracellular-fluid volume decreases reducing venous return and cardiac output until BP returns to normal. Conversely, when BP decreases, the kidney retains salt and water until arterial pressure returns to normal. Thus, pressure natriuresis acts as the key component of the feedback system that normally stabilizes BP and body-fluid volumes. During the early phases of obesity, before loss of nephron function because of glomerular injury, primary sodium retention occurs as a result of increase in renal tubular reabsorption. This may be compensated by renal vasodilation, increased glomerular filtration rate and increased filtered amount of water and electrolytes. As a consequence of an incomplete compensation, however, extracellular-fluid volume is expanded, resulting in a hypertensive adjustment of the pressure natriuresis.8 This resetting of the kidney-fluid apparatus to a hypertensive level is consistent with the model of hypertension because of volume overload. Another significant cause of shift of pressure natriuresis toward higher BP levels in obesity is the possibility of alterations in intrarenal forces caused by histological changes in the renal medulla that may compress the loops of Henle and vasa recta.22
Function of RAS in obesity hypertension
Several studies have shown predominantly high levels of plasma renin activity, plasma angiotensinogen, Ang II and aldosterone values in association with human obesity.23, 24, 25 Hall et al.19 reported a more than twofold increase in plasma renin activity after a 5-week period of high-fat diet administration in conscious dogs. Moreover, fat restriction succeeded in diminishing the values of the RAS components, indicating the possibility that the degree of adiposity might have a direct influence in this BP-regulating system. Under normal conditions, the RAS represents a regulatory mechanism, which prevents extreme variations in arterial pressure evoked by changes in salt intake. Cessation of Ang II formation during high-salt intake results in a decreased rate of BP elevation, as indicated by the leftward shift in the renal function curve closer to the initial BP level.
Despite remarkable volume expansion and sodium retention in obesity, several mechanisms are responsible for the RAS activation. Renin secretion by the kidney seems to be induced by changes in intrarenal physical forces, generating from fat accumulation around and into the renal medulla.22 Owing to the actual histologic changes that cause compression of the medulla, flow rate of the filtrate is diminished at the loop of Henle leading to prolongation of the time given for sodium reabsorption.26 Detection of the decreased amount of sodium, reaching the distal tubular cells, by the macula densa leads to a rise in renin secretion, through tubuloglomerular feedback.
Recent studies elucidated the potent function of the adipose tissue's physiology in BP elevation. Adipose tissue-derived angiotensinogen can enter the circulation. Adipose cells may represent a major site in which all components of the RAS are formed. Renin, Ang II, angiotensinogen and Ang II receptors are found in abundance in adipose mass suggesting that a local tissue Ang system is settled at the adipocytes level.27 The tissue RAS and the circulating RAS are in a state of constant interaction. Angiotensinogen and Ang I and II are locally produced and at the same time are taken up by the cells, in which Ang II receptors are overexpressed. Angiotensinogen production serves as both a cause and effect of adipocyte hypertrophy and leads to elevation of BP through the actions of Ang II, which induce systematic vasoconstriction, direct sodium and water retention and increased aldosterone production. Consequently, Ang II determines a high-salt-sensitive BP condition in obesity as it is produced at high rates and is not suppressed by volume expansion. Another potential mechanism of RAS activation could be a chronic elevation of sympathetic tone, causing renal vasoconstriction and renin-dependent chronic hypertension.
Structural changes in the kidney
Among the multiple causes of renal function impairment, relatively recent data have revealed the implication of a large number of factors provoking changes in renal structure, which, in turn, seem to be a predominant cause of gradual nephron loss, having a great impact on the alteration in pressure natriuresis.22 Physical compression of both kidneys seems to generate from accumulation of adipose tissue around the organs emphasizing the crucial function of visceral obesity in the development of renal disease.28 Deposition of extracellular matrix throughout the renal medulla is greatly expanded and the tissue surrounding the ducts of Bellini at the vascular pole tends to prolapse. Increased numbers of interstitial cells and an increase in material rich in lipids and proteoglycans compresses the renal parenchyma toward the pole of the kidney resulting in the formation of round-shaped, enlarged kidneys in obese subjects.29, 30 Renal compression affects both vascular (mainly the vasa recta) and tubular (the Henle's loops) elements causing activation of the RAS and increased sodium reabsorption.
Renal injury in obesity seems to be directly dependent on body weight, as dietary fat restriction can ameliorate the renal histology.31 The primary histologic features are relatively few lesions of focal-segmental glomerulosclerosis, profound glomerulomegaly because of glomerular hyalinosis and fibrosis, as well as lipid accumulation in the glomeruli and adhesion to Bowman's capsule.32, 33 Some studies suggest that lipid accumulation is a result of alterations in fat metabolism. Whether upregulation of lipogenic enzymes and decreased amounts of lipolytic factors could imply direct lipotoxicity remains an area of active investigation.34 Glomerulomegaly was observed in 100% of renal biopsies in a clinicopathologic study of obesity glomerulopathy. Despite the observed high incidence of glomerulomegaly, glomerular changes in obesity-induced renal injury are incomparable with those of diabetic nephropathy, mainly because of the lower severity of changes in the mesangial space. Other causes of renal injury, apart from high-fat intake, could possibly include overexpression of Ang II with a consequent increase in proliferative factors such as transforming growth factor-β and plasminogen activator inhibitor, high protein diet, as well as hyperinsulinemia giving genesis to insulin growth factors. All of the above may lead to changes in the glomeruli. There is also evidence that SNS response to hypoxia may affect the renal circulation in patients with obstructive sleep apnea, an abnormality often seen in the obese population.4, 30 Hyperfiltration as a result of heightened blood flow to the kidney is always present in obesity, long before glomerulopathy occurs. It is, therefore, the primary cause of gradual sclerosis of the glomeruli wall because of physical stress and eventually of a vicious circle in which nephrons are injured, sodium retention worsens and arterial pressure reaches higher values to maintain sodium balance.
In the absence of diabetes mellitus and hypertension, obesity-related glomerulopathy per se may not promote renal impairment. Clinically significant proteinuria is rare among obese subjects. Microalbuminuria was detected in 25 out of 207 non-diabetic obese patients in a study performed by Valensi et al.35 These findings suggest that obesity-related renal damage should be defined as a special form of focal-segmental glomerulosclerosis slowly progressing to end-stage renal disease.36
Function of hormones
Insulin
Obesity is a state of impaired glucose tolerance, high levels of circulating insulin and reduced sensitivity to the metabolic actions of insulin. This condition is defined as insulin resistance. Whether hyperinsulinemia or insulin resistance is the primary disorder has not yet been clearly delineated. It has been proposed that hyperinsulinemia could compensate for decreased sensitivity to insulin. Normally, insulin exhibits a sodium retaining effect through its direct action on the renal tubules. A potential enhancement of sodium retention because of hyperinsulinemia could lead to a rise in BP. Sechi and Leonardo37 have shown that high dietary salt in normal Sprague–Dawley rats causes a dose-dependent downregulation in the number of insulin receptors in the kidney. However, in genetically predisposed hypertensive rats (SHR), this autoregulative response was blunted. The same effect was also present in fructose-fed hypertensive rats, suggesting a possible function for hyperinsulinemia in the pathogenesis of sodium retention and hypertension in the obese.
Insulin has also been proved to have an acute sympathoexcitatory action in both normotensive and borderline hypertensive subjects, as indicated by increased muscle SNS activity and heightened norepinephrine levels after insulin administration in several studies.38, 39 Insulin release leads to hypoglycemia, which serves as an activator of the SNS. Moreover, vasodilator responses to increased muscular glucose uptake and oxygen demands lead to activation of the baroreceptor reflex and to enhanced muscle SNS activity. Insulin might also have a sympathoexcitatory effect directly on the central nervous system.40 However, in a study by Sakaguchi and Bray,41 insulin injection into the ventromedial hypothalamus produced centrally controlled decreases in sympathetic nerve activity. Another aspect of insulin infusion is the simultaneous depressor effect of peripheral vasodilation42, 43 mediated by a b-adrenergic mechanism.6 Moreover, tachycardia induced by insulin release suggests a withdrawal of parasympathetic tone.
Chronic hyperinsulinemia has been associated with impairment of the vasodilator action of insulin.44 Vasoconstriction in the forearm was reported during insulin infusion in severe insulin resistance.39 This finding suggests that hyperinsulinemia promotes an altered profile of vascular function. Vascular dysfunction seems to be the important factor in understanding the long-term implication of insulin in the causation of hypertension. Whether hypertension is caused by excessive amounts of insulin, resistance to its action or chronically induced trophic vascular effects remains to be uncovered. Finally, insulin resistance has also been exhibited as an effect of heightened sympathetic drive, through b-adrenergic stimulation and/or vasoconstriction with subsequent reduction of muscular blood flow.45, 46
Leptin and neuropeptides
Hyperleptinemia is another possible link between obesity and the development of hypertension. Leptin is a peptide hormone secreted from adipose tissue in direct proportion to adipose tissue mass.47 The amount of leptin secreted from the adipocytes into circulation binds to its short-form receptors and is transported across the blood–brain barrier to the arcuate nucleus, a hypothalamic region of high significance for the transmission of appetite controlling neuropeptides to peripheral tissues. Leptin's primary effects on the hypothalamic neuronal systems include a decrease in food consumption and upregulation of thermogenesis and energy expenditure, through stimulation of sympathetic activity.8, 48 These effects are mediated by two major pathways, one posting a positive-regulating action through expression of the propiomelanocortin-derived peptides—α melanocyte-stimulating hormone (α-MSH)—driven by high leptin levels, and the other exhibiting the opposite actions through the expression of agouti-related peptide and neuropeptide Y (NPY). α-MSH molecules bind to melanocortin 3 and 4 receptors (MC3R and MC4R), act as agonists, stimulate the SNS, increase energy expenditure and activate the hypothalamus–pituitary–adrenal axis. Studies in mice and human beings with leptin deficiency because of mutations of the leptin-expressing gene (ob gene) show high incidence of obesity. The increase in SNS activity develops slowly (hours after infusion of leptin), and in the long term, it may induce sympathetic-mediated hypertension development through raised tubular sodium reabsorption and volume overloading. The cardiovascular actions of leptin are utterly prevented during combined a- and b-adrenergic blockade. In addition, the chronic pressor effects of leptin are supposed to be simultaneously controlled by endothelial nitric oxide (NO) production.49, 50 Deprivation of the endothelium-derived NO markedly promotes BP elevation. On the other hand, agouti-related peptide is an antagonistic ligand of the MC3/MC4 receptor system. Agouti-related peptide is expressed during fasting and leads to increased food consumption. Although obese individuals have high levels of circulating leptin, the expected metabolic actions of leptin leading to reduction in food intake and increased energy expenditure are absent. Selective resistance to the metabolic actions of leptin seems to be present in obesity,49, 50, 51 whereas its action in stimulating the sympathetic tone remains unaltered. An impairment of leptin's transport to the hypothalamus could be a possible reason. Moreover, overexpression of the suppressor of cytokine signaling 3 by high-circulating leptin, which serves as a feedback mechanism, has been suggested to have a function in the pathogenesis of leptin resistance.52
The close relationship of hyperleptinemia with hypertension is further supported by observations obtained in obese leptin-deficiency mice, which are obese, but do not exhibit hypertension. Obesity does not invariably increase BP in mice and probably also in human beings and the arterial pressure response to obesity may depend critically on the underlying genetic and neuroendocrine mechanisms.53 Mutations might reduce the functional activity of leptin. MC4R deficiency abolished the cardiovascular and metabolic actions of leptin in obese MC4R (−/−) mice and possibly in human beings. BP levels have been reported significantly lower in MC4R-deficient subjects than in control subjects.54 Thus, a functional MC4R is essential for the chronic cardiovascular and metabolic actions of leptin.55 MSHs have important function in feeding, energy metabolism and inflammation. Both α- and γ-MSH acutely elevate BP and heart rate through central stimulation of sympathetic nervous outflow.56 This action of α-MSH is mediated by the MC4R, whereas γ-MSH deficiency or disruption of MC3R in rodents leads to salt-sensitive hypertension possibly through a central mechanism. This salt-sensitive hypertension is accompanied by the development of insulin resistance. SNS-stimulating actions of leptin are mainly shown in the kidneys, adrenal glands and brown adipose tissue. SNS stimulation is not the only mediator through which hyperleptinemia leads to cardiovascular reactions. Endothelial dysfunction has also been reported as another important aspect of leptin's effects.57, 58 Leptin is believed to promote direct endothelium toxicity by causing an alteration in the expression of endothelial NO synthase. Finally, it should be considered that perivascular adipose tissue is an additional source of leptin.
NPY is a neurotransmitter also being expressed in the hypothalamic arcuate nucleus at high rates during fasting, having an orexigenic effect combined with reduction in thermogenesis and downregulation of the sympathetic neurons. All the above actions are mediated by the binding of the NPY to receptors Y1–Y6 in the hypothalamus. Normally, NPY expression is suppressed by high leptin levels. In the leptin-resistant state in which leptin's metabolic effects are blunted, NPY should rather be considered overexpressed. NPY released from neural sites by sympathetic activation acts as a vasoconstrictor and could have a function in obesity-related hypertension.59
Adiponectin is an adipose tissue-derived peptide hormone, recently recognized as an energy-balance-regulating molecule. Adiponectin represents a lipolytic factor and a positive regulator of insulin sensitivity. It also prevents the atherogenetic process by inhibiting foam-cell formation. Adiponectin interacts with peripheral receptors AdipoR1 and AdipoR2, which have also been detected at the hypothalamic arcuate nucleus.60 Adiposity is characterized by adiponectin deficiency, which leads to increased thermogenesis, although it does not affect food intake. Plasma adiponectin levels are inversely related to insulin resistance.61 Adiponectin knockout mice show severe diet-induced insulin resistance and atherogenesis. These data suggest that hypoadiponectinemia is one of the factors responsible for cardiovascular symptom clustering in obesity.
Ghrelin is an orexigenic hormone secreted by the oxyntic cells of the stomach. Its secretion is augmented during fasting. Ghrelin is the endogenous agonist of growth hormone receptor in the hypothalamus.48 Although ghrelin production is known to be suppressed in adiposity, high levels of ghrelin have been reported in obese hypertensive patients. Weight loss has been shown to induce an increase in ghrelin levels.62 Moreover, it has been shown that ghrelin may decrease BP, improve endothelial function and inhibit vascular oxidative stress in spontaneously hypertensive rats.63
The endocannabinoids represent polyunsaturated fatty-acid derivatives, which take part in a large neuromodulatory signaling system for the control of balance between food intake and satiation. It consists of the endogenous lipids cannabinoids produced in the cell membrane of post-junctional cells and two types of G-coupled transmembrane receptors, CB1 and CB2. CB1 receptors mediate energy homeostasis, food consumption, lipid and glucose metabolism through expression of their molecule in a large number of central and peripheral tissues.64 They are localized at several sites in the central nervous system, and when activated, lead to appetite stimulation, which in the long term is able to establish a vicious circle of overactive endocannabinoid system-high-caloric intake obesity. CB1 receptors are also found at several peripheral sites such as adipose tissue, liver, skeletal muscles and vascular endothelial and smooth muscle cells. Activation of the endocannabinoid system has been implicated in CB1-mediated hypotension associated with hemorrhagic, septic, cardiogenic shock and advanced liver cirrhosis.64 Endocannabinoid system overactivity may induce hepatic lipogenesis through expression of the transcriptor factor sterol regulatory element-binding protein-1c and its target enzymes acetyl-CoA carboxylase and fatty-acid synthase reducing muscular oxygen consumption and glucose uptake, downregulating adiponectin secretion in the adipose tissue and finally affecting the transmission of satiety signals from the gastrointestinal tract. It is, therefore, obvious that endocannabinoid system stimulation represents a source of multiple risk factors clustering, including visceral obesity, insulin resistance, glucose intolerance, hyperlipidemia and reduced adiponectin. Blockade of the CB1 receptors could have been a novel therapeutic approach to reduce the total cardiometabolic risk, but psychiatric adverse effects represent a major concern. A recent study showed that the reduction in body weight achieved by the CB1 blocker rimonabant was mainly because of increased energy expenditure than reduced food intake.65 A meta-analysis of the BP effect of rimonabant reported that in the subgroup of patients with hypertension, there was a modest hypotensive effect over placebo and possibly suggests that a pressor effect of rimonabant may limit the BP reduction that should be achieved through weight loss.66 There was no weight-independent effect of rimonabant on BP and the degree of reduction was lower than that expected by the weight loss.67
Corticosteroids
The hypothesis that the hypothalamic–pituitary–adrenal axis could possibly take part in the pathophysiology of obesity-related hypertension is supported by similarities in phenotype between Cushing's syndrome and visceral obesity-associated disease. Glucocorticoids increase food intake, reduce energy expenditure and they promote insulin resistance, fat accumulation68 and hypertension.69, 70 Many rodent models of obesity, which are characterized by hypercorticosteronemia with weight gain, displayed body weight normalization after adrenalectomy and reinstated by glucocorticoid replacement. Although plasma glucocorticoid levels are normal in human idiopathic obesity,71 it has been proposed that intra-adipose glucocorticoid action is increased. Obese individuals have selectively increased adipose levels of 11b-hydroxysteroid dehydrogenase-1,72, 73 an enzyme that regenerates active cortisol from inactive 11-keto forms. The aP2-HSD1 mice, with relative transgenic overexpression of this enzyme in fat cells, develop visceral obesity with insulin resistance, dyslipidemia and hypertension. These mice have increased sensitivity to dietary salt and increased plasma levels of angiotensinogen, Ang II and aldosterone. Hypertension in aP2-HSD1 mice was abolished by selective Ang II receptor antagonists at a low dose.74 These findings propose that a local activation of the adipose glucocorticoid action induce an activation of the RAS, which mediates a salt-sensitive form of hypertension in obesity.
Endothelial dysfunction and changes in vascular structure
There is increasing evidence supporting the significance of vascular endothelial dysfunction in the pathogenesis of hypertension. Obesity represents a state of inflammation (vascular and systemic) that can cause endothelial dysfunction. Insulin resistance, low levels of adiponectin, high plasma leptin, increased levels of plasma glucose and FFAs are considered as indices of an inflammation compatible profile. A series of insulin signaling pathways are instructed to function in favor of inflammation and uncontrolled endothelial growth causing endothelial dysfunction and finally hypertension.
NO, which derives from the vascular endothelium, promotes vasodilation, whereas at the same time protects from inflammation and platelet aggregation. Insulin-dependent phosphoinositide 3-kinase activation normally leads to phosphorylation of endothelial NO synthase and a subsequent increase in NO production. In the presence of insulin resistance, this pathway is downregulated leading to impairment of NO synthesis, whereas hyperinsulinemia increases vasoconstrictor endothelin-1 levels,75, 76 as the mitogen-activated protein-kinase pathway mediating endothelin-1 production remains unaltered. As a result, an imbalance between vasodilator and vasoconstrictor actions in the vascular endothelium is provoked. Another mitogen-activated protein -kinase controlled pathway is the production of adhesion molecules, vascular cell adhesion molecule-1, inter-cellular adhesion molecule-1 and E-selectin by the endothelial cells, promoting monocyte adhesion to the vascular wall. Similar mechanisms are brought into operation because of elevated serum levels of glucose and FFAs.
A variety of biologically active derivatives generate from adipose cells, including reactive oxygen species, proinflammatory and inflammatory molecules (interleukin-1β, interleukin-6, tumor necrosis factor-α, C-reactive protein), angiogenetic factors (vascular endothelial growth factor),77 hemostasis modulating compounds (plasminogen activator inhibitor-1, thromboxane A2) and acute phase reaction proteins (serum amyloid A proteins, C-reactive protein). Activation of nuclear factor κ light-chain enhancer of activated B cells (NF-κB) and IκB kinase78 because of fat accumulation is of great significance for the establishment of a proinflammatory and prothrombotic state, indicating the presence of altered vascular function that predisposes to the development of hypertension.
Earlier studies had also reported that the mean intima media thickness of the internal carotid arteries was increased from the lowest to the highest quartile of BMI. Intima media thickness was found significantly higher in obese compared with normal weight subjects in groups of patients matched for age, sex and ambulatory BP levels.14 These results suggest that obesity per se may be a major risk factor for carotid atherosclerosis. Intima media thickness in obese subjects was independently associated with fasting serum glucose levels, suggesting that obesity-induced hyperglycemia is an important predictor of carotid atherosclerosis. As atherosclerosis progresses, large arteries become stiffer. Increased pulse wave velocity and arterial stiffness may contribute to obesity hypertension. Long-term sympathetic and RAS activation leads to small artery vasoconstriction and remodeling, which increases wall to lumen ratio and may act synergistically with large artery damage to raise BP. In obese patients, aortic pulse wave velocity rises not only because of aortic stiffening, but also possibly because of remodeling of the small and medium sizes arteries. As a result of these changes, the amount of the reflected pressure waves in the periphery arrives back earlier at the central arteries augmenting systolic BP.
Therapeutic approaches for obesity-induced hypertension
Current guidelines suggest lifestyle modifications and reduction in body weight for all obese hypertensive patients.3 A low-caloric diet reducing total-caloric intake by 500–1000 calories per day should be proposed. The aim of this approach should be a modest weight loss (10% of body weight over 6 months) rather than an extreme dietary goal that is usually followed by quick weight regain. Modest restriction of salt intake, low intake of saturated fats and cholesterol with increased consumption of fruits, vegetables and whole grains is recommended.79, 80 Moderate physical activity should accompany every dietary program.81 Supplementary behavioral therapy including self-monitoring, psychological support and stress management could also be helpful. Only a few patients are able to achieve long-term weight reductions with life style interventions. Surgical approaches should be reserved for morbid obese patients (BMI⩾40 kg m–2) with hypertension and other major comorbidities.80 In the prospective, controlled Swedish Obese Subjects study,82 subjects who underwent bariatric surgery had lower overall mortality and long-term weight loss, during an average of 10.9 years of follow-up, compared with a matched control group that received conventional treatment. The surgery group had lower 10-year incidence rates of diabetes, hypertriglyceridemia and hyperuricemia, but not of hypertension than the control group. These findings may suggest that weight loss achieved by bariatric surgery may not result in long-term BP reduction.
Pharmacological treatment for obesity is recommended in selected patients severely affected by obesity and its sequelae. Available weight reduction drugs include orlistat, a fat-absorption (lipase) inhibitor and sibutramine, an appetite-suppressing drug with central action (norepinephrine and serotonin reuptake inhibition).81 Orlistat reduced BP83 with side effect being the reduction of fat-soluble vitamins absorption. Sibutramine was reported to induce tachycardia and probably increased BP83 as a result of the interaction between peripheral and central nervous system effects, peripheral stimulatory effects and inhibitory clonidine-like actions on the central nervous system.84 Rimonabant, a novel CB1-receptor inhibitor at the CNS and peripheral tissues, has been associated with increased rates of anxiety and depression disorders, and the European Medicines Agency has recently recommended the suspension of the marketing authorization for the treatment of obesity.
Although all categories of antihypertensive drugs could be administered in obese hypertensives, there are some limitations (Figure 2). Diuretic drugs especially in high doses present significant side effects such as hyperglycemia, hyperlipidemia and hyperuricemia.85 Administration of these drugs in an obese patient would magnify these disorders. β-blockers may cause impaired lipid and glucose metabolism and should not be used as first choice therapy in obese hypertensive patients unless required by specific indications.86, 87 However, these effects seem to be less pronounced with the new vasodilating β-blockers such as carvedilol and nebivolol.88 First choice drugs for the obesity-induced hypertension are the Ang receptor antagonists and the ACE-inhibitors both associated with a lower incidence of diabetes85 and favorable effects on left ventricular hypertrophy and nephropathy. Ca++ channels antagonists can be used alternatively or can be added to the treatment with Ang receptor antagonists and the ACE inhibitors, if BP is resistant to monotherapy.89 Calcium antagonists are metabolically neutral drugs and exhibit favorable actions on target organ damage. Thiazide diuretics at low doses may be added if BP is not controlled, although they might have some adverse effect on insulin resistance and new onset of diabetes.85 Other comorbidities including dislipidemia and diabetes should also be appropriately treated to reduce total cardiovascular risk in obese patients.

Target BP⩽130/85 mm Hg if >3 risk factors or diabetes or metabolic syndrome or renal failure or target organ damage exist, otherwise target BP⩽140/90 mm Hg. BP, blood pressure; ACEI, ace-converting enzyme inhibitors; ARB, angiotensin-II receptor blockers; CCB, calcium channel blockers; HCZ, hydrochlorothiazide. Stage II hypertension with multiple risk factors or established cardiovascular disease and stage III hypertension with immediate drug treatment.
Obesity should be considered as a chronic medical condition, which is likely to require long-term treatment. The presence of obesity-related outcomes such as hypertension and diabetes may be indications for more aggressive therapy. Multiple therapeutic approaches may be necessary to achieve the desired goal. Understanding of the mechanisms associated with obesity-related hypertension is essential for refined treatment strategies. Future research on the mechanisms of obesity-related hypertension is thus needed, and may serve as the basis for more effective and well-tolerated therapies.
References
Kotsis V, Stabouli S, Bouldin M, Low A, Toumanidis S, Zakopoulos N . Impact of obesity on 24-h ambulatory blood pressure and hypertension. Hypertension 2005; 45: 602–607.
Stabouli S, Kotsis V, Papamichael C, Constantopoulos A, Zakopoulos N . Adolescent obesity is associated with high ambulatory blood pressure and increased carotid intimal medial thickness. J Ped 2005; 147: 651–656.
Mancia G, De Backer G, Dominiczak A, Cifkova R, Fagard R, Germano G, Grassi G, Heagerty AM, Kjeldsen SE, Laurent S, Narkiewicz K, Ruilope L, Rynkiewicz A, Schmieder RE, Boudier HA, Zanchetti A, Vahanian A, Camm J, De Caterina R, Dean V, Dickstein K, Filippatos G, Funck-Brentano C, Hellemans I, Kristensen SD, McGregor K, Sechtem U, Silber S, Tendera M, Widimsky P, Zamorano JL, Erdine S, Kiowski W, Agabiti-Rosei E, Ambrosioni E, Lindholm LH, Viigimaa M, Adamopoulos S, Agabiti-Rosei E, Ambrosioni E, Bertomeu V, Clement D, Erdine S, Farsang C, Gaita D, Lip G, Mallion JM, Manolis AJ, Nilsson PM, O’Brien E, Ponikowski P, Redon J, Ruschitzka F, Tamargo J, van Zwieten P, Waeber B, Williams B, Management of Arterial Hypertension of the European Society of Hypertension; European Society of Cardiology. Guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens 2007; 25: 1105–1187.
Grassi G, Seravalle G, Cattaneo BM, Bolla G, Lafranchi A, Colombo M, Giannattasio C, Brunani A, Cavagnini F, Mancia G . Sympathetic activation in obese normotensive subjects. Hypertension 1995; 25: 560–563.
Landsberg L, Krieger DR . Obesity, metabolism, and the sympathetic nervous system. Am J Hypertens 1989; 2: 1255–1325.
Rocchini AP, Yang IQ, Gokee A . Hypertension and insulin resistance are not directly related in obese dogs. Hypertension 2004; 43: 1011–1016.
Coatmellec-Taglioni G, Ribière C . Factors that influence the risk of hypertension in obese individuals. Curr Opin Nephrol Hypertens 2003; 12: 305–308.
Hall JE, Brands MW, Hildebrandt DA, Kuo J, Fitzgerald S . Role of sympathetic nervous system and neuropeptides in obesity hypertension. Braz J Med Biol Res 2000; 33: 605–618.
Wofford MR, Anderson Jr DC, Brown CA, Jones DW, Miller ME, Hall JE . Antihypertensive effect of alpha- and beta-adrenergic blockade in obese and lean hypertensive subjects. Am J Hypertens 2001; 14: 694–698.
Hall JE, Louis K . Dahl Memorial Lecture. Renal and cardiovascular mechanisms of hypertension in obesity. Hypertension 1994; 23: 381–394.
Narkiewicz K, Van de Borne P, Cooley R, Dyken M, Somers V . Sympathetic activity in obese subjects with and without obstructive sleep apnea. Circulation 1998; 98: 772–776.
Parati G, Lombardi C, Narkiewicz K . Sleep apnea: epidemiology, pathophysiology, and relation to cardiovascular risk. Am J Physiol Regul Integr Comp Physiol 2007; 293: R1671–R1683.
Weyer C, Prattley RE, Snitker S, Spraul M, Ravussin E, Tataranni PA . Ethnic differences in insulinemia and sympathetic tone as links between obesity and blood pressure. Hypertension 2000; 36: 531–537.
Kotsis V, Stabouli S, Papamichael C, Zakopoulos N . Impact of obesity in intima media thickness of carotid arteries. Obesity 2006; 14: 1708–1715.
Stepniakowski KT, Goodfriend TL, Egan BM . Fatty acids enhance vascular a-adrenergic sensitivity. Hypertension 1995; 25: 774–778.
Oishi K, Zheng B, Kuo JF . Inhibition of Na, K-ATPase and sodium pump by protein kinase C regulators sphingosine, lysophosphatidylcholine and oleic acid. J Biol Chem 1990; 265: 70–75.
Khan WA, Blobe G, Halpern A, Taylor W, Wetsel WC, Burns D, Loomis C, Hannun Y . Selective regulation of protein kinase C isoenzymes by oleic acid in human platelets. J Biol Chem 1993; 268: 5063–5068.
Ordway R, Singer J, Walsh J . Direct regulation of ion channels by fatty acids. Trends Neurosci 1991; 14: 96–100.
Hall JE, Brands MW, Dixon WN, Smith MJ . Obesity-induced hypertension. Renal function and systemic hemodynamics. Hypertension 1993; 22: 292–299.
Taddei S, Virdis A, Mattei P, Favilla S, Salvetti A . Angiotensin II and sympathetic activity in sodium-restricted essential hypertension. Hypertension 1995; 25: 595–601.
Guyton AC . The surprising kidney-fluid mechanism for pressure control-its infinite gain!. Hypertension 1990; 16: 725–730.
Hall JE . Mechanisms of abnormal sodium handling in obesity hypertension. Am J Hypertens 1997; 10: S49–S55.
Ruano M, Silvestre V, Castro R, García-Lescún MC, Rodríguez A, Marco A, García-Blanch G . Morbid obesity, hypertensive disease and the renin-angiotensin-aldosterone axis. Obes Surg 2005; 15: 670–676.
Kidambi S, Kotchen J, Grim C, Raff H, Mao J, Singh R, Kotchen T . Association of adrenal steroids with hypertension and the metabolic syndrome in blacks. Hypertension 2007; 49: 704–711.
Massièra F, Bloch-Faure M, Ceiler D, Murakami K, Fukamizu A, Gasc JM, Quignard-Boulangé A, Neyrel R . Adipose angiotensinogen is involved in adipose tissue growth and blood pressure regulation. FASEB J 2001; 15: 2727–2729.
Rocchini AP . Obesity hypertension. Am J Hypertens 2002; 15: 50S–52S.
Campbell DJ . Circulating and tissue angiotensin systems. J Clin Invest 1987; 79: 1–6.
Hall JE, Crook ED, Jones DW, Wolford MR, Dubbert PM . Mechanisms of obesity-associated cardiovascular and renal disease. Am J Med Sci 2002; 324: 127–137.
Dwyer TM, Bigler SA, Moore NA, Carroll JF, Hall JE . The altered structure of renal papillary outflow tracts in obesity. Ultrastruct Pathol 2000; 24: 251–257.
Kambham N, Markowitz GS, Valeri AM, D’Agati VD . Obesity-related glomerulopathy: an emerging epidemic. Kidney Int 2001; 59: 1498–1509.
Deji N, Kume S, Araki S, Soumura M, Sugimoto T, Isshiki K, Chin-Kanasaki M, Chin-Kanasaki M, Sakaguchi M, Koya D, Haneda M, Kashiwagi A, Uzu T . Structural and functional changes in the kidneys of high-fat diet-induced obese mice. Am J Physiol Renal Physiol 2009; 296: F118–F126.
Chalmers L, Kaskel FJ, Bamgbola O . The role of obesity and its bioclinical correlates in the progression of chronic kidney disease. Adv Chronic Kidney Dis 2006; 13: 352–364.
Hall JE, Brands MW, Henegar JR . Mechanisms of hypertension and kidney disease in obesity. Ann N Y Acad Sci 2006; 892: 91–107.
Kume S, Uzu T, Araki S, Sugimoto T, Isshiki K . Role of altered renal lipid metabolism in the development of renal injury induced by a high-fat diet. J Am Soc Nephrol 2007; 18: 2715–2723.
Valensi P, Assayag M, Busby M . Microalbuminuria in obese patients with or without hypertension. Int J Obes Relat Metab Disord 1996; 20: 574–579.
Praga M, Hernández E, Morales E, Campos AP, Valero MA, Martinez MA, Léon M . Clinical features and long-term outcome of obesity associated focal segmental glomerulosclerosis. Nephrol Dial Transplant 2001; 16: 1790–1798.
Sechi M, Leonardo A . Mechanisms of insulin resistance in rat models of hypertension and their relationships with salt sensitivity. J Hypertens 1999; 17: 1229–1237.
Anderson EA, Balou TW, Hoffman RP, Sinkey CA, Mark AL . Insulin increases sympathetic activity but not blood pressure in borderline hypertensive humans. Hypertension 1992; 19: 621–627.
Gudbjörnsdottir S, Elam M, Sellgren J, Anderson EA . Insulin increases forearm vascular resistance in obese, insulin-resistant hypertensives. J Hypertens 1996; 14: 91–97.
Landsberg L . Diet, obesity and hypertension: a hypothesis involving insulin, the sympathetic nervous system, and adaptive thermogenesis. Q J Med 1986; 236: 1081–1090.
Sakaguchi T, Bray GA . Intrahypothalamic injection of insulin decreases firing rate of sympathetic nerves. Proc Natl Acad Sci USA 1987; 84: 2012–2014.
Hall JE . Hyperinsulinemia: a link between obesity and hypertension? Kidney Int 1993; 43: 1402–1417.
Creager MA, Liang CS, Coffman JD . Beta adrenergic-mediated vasodilator response to insulin in the human forearm. Am Soc Pharm Exper Ther 1985; 235: 709–714.
Hall JE, Brands MW, Zappe DH, Dixon WN, Mizelle L, Reinhart GA, Hildebrandt DA . Hemodynamic and renal responses to chronic hyperinsulinemia in obese, insulin-resistant dogs. Hypertension 1995; 25: 994–1002.
Julius S, Jamerson K . Sympathetics, insulin resistance and coronary risk in hypertension: the ‘chicken-and-egg’ question. J Hypertens 1994; 12: 495–502.
Masuo K, Mikami H, Ogihara T, Tuck ML . Weight gain-induced blood pressure elevation. Hypertension 2000; 35: 1135–1140.
DeCourten M, Zimmet P, Hodge A, Collins V, Nicolson M, Staten M, Dowse G, Alberti KG . Hyperleptinemia: the missing link in the metabolic syndrome? Diabet Med 1997; 14: 200–208.
Wynne K, Stanley S, McGowan B, Bloom S . Appetite control. J Endocrinol 2005; 184: 291–318.
Engeli S, Sharma A . Emerging concepts in the pathophysiology and treatment of obesity-associated hypertension. Curr Opin Cardiol 2002; 17: 355–359.
Kuo JJ, Jones OB, Hall JE . Inhibition of NO synthesis enhances chronic cardiovascular and renal actions of leptin. Hypertension 2001; 37: 670–676.
Correia ML, Haynes WG, Rahmouni K, Morgan DA, Sivitz WI, Mark AL . The concept of selective leptin resistance: evidence from agouti yellow obese mice. Diabetes 2002; 51: 439–442.
Denis RG, Bing C, Brocklehurst S, Harrold JA, Vernor RG, Williams G . Diurnal changes in hypothalamic neuropeptide and SOCS-3 expression: effects of lactation and relationship with serum leptin and food intake. J Endocrinol 2004; 183: 173–181.
Mark AL, Shaffer RA, Correia ML, Morgan DA, Sigmund CD, Haynes WG . Contrasting blood pressure effects of obesity in leptin-deficient ob/ob mice and agouti yellow obese mice. J Hypertens 1999; 17: 1949–1953.
Greenfield JR, Miller JW, Keogh JM, Henning E, Satterwhite JH, Cameron GS, Astruc B, Mayer JP, Brage S, See TC, Lomas DJ, O’Rahilly S, Farooqi IS . Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med 2009; 360: 44–52.
Tallam LS, da Silva AA, Hall JE . Melanocortin-4 receptor mediates chronic cardiovascular and metabolic actions of leptin. Hypertension 2006; 48: 58–64.
Humphreys MH . Cardiovascular and renal actions of melanocyte-stimulating hormone peptides. Curr Opin Nephrol Hypertens 2007; 16: 32–38.
Knudson JD, Payne GA, Borbouse L, Tune JD . Leptin and mechanisms of endothelial dysfunction and cardiovascular disease. Curr Hypertens Rep 2008; 10: 434–439.
Korda M, Kubant R, Patton S, Malinski T . Leptin-induced endothelial dysfunction in obesity. Am J Physiol Heart Circ Physiol 2008; 295: H1514–H1521.
Lundberg JM, Pernow J, Franco-Cereceda A, Rudehill A . Effects of antihypertensive drugs on sympathetic vascular control in relation to NPY. J Cardiovasc Pharmacol 1987; 10 (Suppl 12): S51–S68.
Guillod-Maximin E, Roy AF, Vacher CM, Aubourg A, Bailleux V, Lossignol A, Penicaud L, Parquet M, Taonis M . Adiponectin receptors are expressed in hypothalamus and colocalized with POMC and NPY in rodent arcuate neurons. J Endocrinol 2009; 200: 93–105.
Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, Tataranni PA . Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocr Metab 2001; 86: 1930–1935.
Hansen TK, Dall R, Hosoda H, Kojima M, Kangawa K, Christiansen JS, Jørgensen JO . Weight loss increases circulating levels of ghrelin in human obesity. Clin Endocrinol (Oxf) 2002; 56: 203–206.
Kawczynska-Drozdz A, Olszanecki R, Jawien J, Brzozowski T, Pawlik WW, Korbut R, Guzik TJ . Ghrelin inhibits vascular superoxide production in spontaneously hypertensive rats. Am J Hypertens 2006; 19: 764–767.
Herling AW, Kilp S, Elvert R, Haschke G, Kramer W . Increased energy expenditure contributes more to the body weight-reducing effect of rimonabant than reduced food intake in Candy-fet wistar rats. Endocrinology 2008; 149: 2557–2566.
Pacher P, Mukhopadhyay P, Mohanraj R, Godlewski G, Bátkai S, Kunos G . Modulation of the endocannabinoid system in cardiovascular disease. Hypertension 2008; 52: 601–607.
Ruilope LM, Despres JP, Scheen A, Pi-Sunyer X, Mancia G, Zanchetti A, Van Gaal L . Effect of rimonabant on blood pressure in overweight/obese patients with/without co-morbidities: analysis of pooled RIO study results. J Hypertens 2008; 26: 357–367.
Neter JE, Stam BE, Kok FJ, Grobbee DE, Geleijnse JM . Influence of weight reduction on blood pressure: a meta-analysis of randomized controlled trials. Hypertension 2003; 42: 878–884.
Kellendonk C, Eiden S, Kretz O, Schutz G, Schmidt I, Tronche F, Simon E . Inactivation of the GR in the nervous system affects energyaccumulation. Endocrinology 2002; 143: 2333–2340.
Saruta T . Mechanism of glucocorticoid-induced hypertension. Hypertens Res 1996; 19: 1–8.
Whitworth JA, Schyvens CG, Zhang Y, Mangos GJ, Kelly JJ . Glucocorticoid-induced hypertension: from mouse to man. Clin Exp Pharmacol Physiol 2001; 28: 993–996.
Flier JS . Obesity wars: molecular progress confronts an expanding epidemic. Cell 2004; 116: 337–350.
Michailidou Z, Coll AP, Kenyon CJ, Morton NM, O’Rahilly S, Secki JR, Chapman KE . Peripheral mechanisms contributing to the glucocorticoid hypersensitivity in POMC null mice treated with corticosterone. J Endocrinol 2007; 194: 161–170.
Lindsay RS, Wake DJ, Nair S, Bunt J, Livingstone DE, Permana PA, Tataranni PA, Walker BR . Subcutaneous adipose 11b-hydroxysteroid dehydrogenase type 1 activity and messenger ribonucleic acid levels are associated with adiposity and insulinemia in Pima Indians and Caucasians. J Clin Endocrinol Metabol 2003; 88: 2738–2744.
Masuzaki H, Yamamoto H, Kenyon CJ, Elmquist JK, Morton NM, Paterson JM, Shinyama H, Sharp MGF, Fleming S, Mullins JJ, Seckl JR, Flier JS . Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J Clin Invest 2003; 112: 83–90.
Kim J, Montagnani M, Koh KK, Quon MJ . Reciprocal relationships between insulin resistance and endothelial dysfunction. Circulation 2006; 113: 1888–1904.
Montagnani M, Quon MJ . Insulin action in vascular endothelial potential mechanisms linking insulin resistance with hypertension. Diab Obes Metabol 2000; 2: 285–292.
Ledoux S, Quequiner J, Msika S, Calderari S, Rufat P, Gasc JM, Corvol P, Larger E . Angiogenesis associated with visceral and subcutaneous adipose tissue in severe human obesity. Diabetes 2008; 57: 3247–3257.
Kim F, Pham M, Maloney E, Rizzo NO, Morton GJ, Wisse BE, Kirk EA, Chait A, Schwartz MW . Vascular inflammation, insulin resistance and reduced nitric oxide production percede the onset of peripheral insulin resistance. Arterioscler Thromb Vasc Biol 2008; 28: 1982–1988.
Mancia G, Bousquet P, Elghozi JL, Esler M, Grassi G, Julius S, Reid J, Van Zwieten PA . The sympathetic nervous system and the metabolic syndrome. J Hypertens 2007; 25: 909–920.
Clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults-the evidence report. National Institutes of Health. Obes Res 1998; 2 (suppl 6): 51S–209S.
Krauss RM, Eckel RH, Howard B, Appel LJ, Daniels SR, Deckelbaum RJ, Erdman Jr JW, Kris-Etherton P, Goldberg IJ, Kotchen TA, Lichtenstein AH, Mitch WE, Mullis R, Robinson K, Wylie-Rosett J, St Jeor S, Suttie J, Tribble DL, Bazzarre TL . AHA DietaryGuidelines: revision 2000: a statement for healthcare professionals from the Nutrition Committee of the American Heart Association. Circulation 2000; 102: 2284–2299.
Sjöström L, Narbro K, Sjöström CD, Karason K, Larsson B, Wedel H, Lystig T, Sullivan M, Bouchard C, Carlsson B, Bengtsson C, Dahlgren S, Gummesson A, Jacobson P, Karlsson J, Lindroos A-K, Lönroth H, Näslund I, Olbers T, Stenlöf K, Torgerson J, Ågren G, Carlsson LMS, for the Swedish Obese Subjects Study. Effects of bariatric surgery on mortality in Swedish obese subjects. N Engl J Med 2007; 357: 741–752.
Siebenhofer A, Horvath K, Jeitler K, Berghold A, Stich AK, Matyas E, Pignitter N, Siering U . Long-term effects of weight-reducing drugs in hypertensive patients. Cochrane Database Syst Rev 2009; Jul 8: CD007654.
Birkenfeld A, Schroeder C, Boschmann M, Tank J, Franke G, Luft F, Biaggioni I, Sharma A, Jordan J . Paradoxical effect of sibutramine on autonomic cardiovascular regulation. Circulation 2002; 106: 2459–2465.
Mancia G, Grassi G, Zanchetti A . New-onset diabetes and antihypertensive drugs. J Hypertens 2006; 24: 3–10.
Pischon T, Sharma AM . Use of beta-blockers in obesity hypertension: potential role of weight gain. Obes Rev 2001; 2: 275–280.
Jacob S, Rett K, Henriksen EJ . Antihypertensive therapy and insulin sensitivity: do we have to redefine the role of beta-blocking agents? Am J Hypertens 1998; 11: 1258–1265.
Kaiser T, Heise T, Nosek L, Eckers U, Sawicki PT . Influence of nebivolol and enalapril on metabolic parameters and arterial stiffness in hypertensive type 2 diabetic patients. J Hypertens 2006; 24: 1397–1403.
Pepine CJ, Handberg EM, Cooper-DeHoff RM, Marks RG, Kowey P, Messerli FH, Mancia G, Cangiano JL, Garcia-Barreto D, Keltai M, Erdine S, Bristol HA, Kolb HR, Bakris GL, Cohen JD, Parmley WW, INVEST Investigators. A calcium antagonist vs a non-calcium antagonisthypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290: 2805–2816.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Kotsis, V., Stabouli, S., Papakatsika, S. et al. Mechanisms of obesity-induced hypertension. Hypertens Res 33, 386–393 (2010). https://doi.org/10.1038/hr.2010.9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2010.9
Keywords
This article is cited by
-
Combined versus independent effects of exercise training and intermittent fasting on body composition and cardiometabolic health in adults: a systematic review and meta-analysis
Nutrition Journal (2024)
-
The ongoing impact of obesity on childhood hypertension
Pediatric Nephrology (2024)
-
Additive interactions between obesity and insulin resistance on hypertension in a Chinese rural population
BMC Public Health (2023)
-
Don’t miss the isolated diastolic hypertension
Hypertension Research (2023)
-
Mixed effects logistic regression analysis of blood pressure among Ghanaians and associated risk factors
Scientific Reports (2023)
