
Abstract
Adrenal adenomas producing both aldosterone and cortisol (A/CPAs) have been described in only a few cases. Correct subtype classification is necessary for making therapeutic decisions in primary aldosteronism (PA). Therefore, we studied in detail the clinical, hormonal and histological features of this entity in two patients with A/CPAs. We describe two patients with A/CPA and present their endocrine evaluations at baseline, after suppression with fludrocortisone and dexamethasone, after therapy with spironolactone and after unilateral adrenalectomy. Moreover, the expression of corticotropin (MC2R) and angiotensin II type 1 (AT1R) receptors and 17α-hydroxylase in the tumors of these two patients was analyzed by immunohistochemistry. Aldosterone, 18-hydroxycorticosterone (18-OH-B) and 18-hydroxycortisol (18-OH-F) were not suppressible with fludrocortisone in either patient and were partly suppressible with dexamethasone in one of the patients. Adrenal insufficiency developed in both patients after operation and lasted for more than 6 months. Aldosterone and hybrid corticosteroids returned to normal 8 weeks after adrenalectomy. In both cases, immunostaining showed weak expression of AT1R and MC2R but strong expression of 17α-hydroxylase. The most common germline mutations in the aldosterone synthase gene and the aldosterone synthase/11β-hydroxylase hybrid gene were absent. These two cases document the fact that sporadic A/CPA is a subtype of PA. The presence of an A/CPA should be considered if a patient has both PA and hypercortisolism.
Similar content being viewed by others

Introduction
Primary aldosteronism (PA) is the consequence of an overproduction of aldosterone in the adrenal gland and is one of the major causes of secondary hypertension.1, 2, 3, 4 PA is characterized by suppressed plasma renin concentration, inadequately high secretion of aldosterone and an elevated aldosterone-to-renin ratio. The overproduction of this mineralocorticoid derives from diffuse or micronodular hyperplasia of the adrenal cortex or from unilateral aldosterone-producing adrenocortical adenomas (APA). Rare forms include mainly the three defined familial forms of hyperaldosteronism and adrenocortical carcinoma.5, 6
In addition, approximately 34 cases of APA with hypersecretion of cortisol (A/CPA) have been described in the literature.7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 Here, we report comprehensive data in two new cases, including clinical presentation, endocrine function tests and immunohistochemical findings in the tumors of both patients, who suffered from both Conn's and Cushing's syndromes because of aldosterone and cortisol co-hypersecretion by sporadic adrenocortical tumors.
Methods
Case reports
Patient 1
A 57-year-old postmenopausal female who had suffered from a hypertensive crisis with hypokalemia was referred to our outpatient clinic for further examination. The patient reported nocturia, dry mouth, sporadic episodes of vertigo and uneasiness. She was obese with a height of 1.53 m, weight of 74.2 kg and a body mass index of 32 kg m−2. Her blood pressure was 200/99 mm Hg and her pulse rate 82 b.p.m. More characteristics are given in Table 1. She exhibited features of Cushing's syndrome, including truncal obesity, muscle weakness and hirsutism, along with a mild atrophy of the skin and osteoporosis. Her laboratory data showed high normal sodium (145 mmol l−1) and decreased potassium (3.3 mmol l−1) levels. Her platelet count was rather high (573 × 10ł μl). Analysis of a 24-h urine sample showed elevated secretion of aldosterone-18-glucuronide (17.6 μg per 24 h, normal 3.5–17.5 μg per 24 h) and tetrahydroaldosterone (171.4 μg per 24 h, normal 10–70 μg per 24 h). Further endocrine workup showed suppressed levels of corticotropin (ACTH), dehydroepiandrosterone-sulfate (DHEAS) and renin, along with excessive levels of cortisol and aldosterone (Tables 2 and 3). Notably, there was no suppression of aldosterone by fludrocortisone or cortisol by medium-dose dexamethasone, and levels of 18-hydroxycortisol (18-OH-F) remained elevated during testing (Table 3). A renal artery stenosis was considered unlikely by color duplex ultrasound. Subsequent MRI scans showed a tumor in the left adrenal gland, consistent with an adrenocortical adenoma. She had no family history of adrenal tumors or arterial hypertension. She had, however, undergone an operation on the neck for a thyroid nodule that turned out to be papillary thyroid cancer (pT2(m), pN0(0/2), MX, L0, V0, R0).
After pretreatment with 25 mg per day spironolactone for 3 weeks and 50 mg per day spironolactone for another 3 weeks, retroperitoneoscopic adrenalectomy was performed and the affected adrenal gland was removed. The latter procedure was followed by treatment with hydrocortisone that was gradually reduced over 12 months. The blood pressure had returned to normal within 1 year without antihypertensive drugs.
Patient 2
A 49-year-old female was referred to our outpatient clinic for further investigation of PA. Two months before admission, she had been hospitalized because of a hypertensive crisis. A pheochromocytoma was excluded by normal plasma metanephrines and normetanephrines in subsequent investigations. An MRI study excluded renal artery stenosis and showed an adrenal tumor, consistent with a cortical adenoma (Table 1).
The patient suffered from depressive episodes. She had a 2-year history of elevated blood pressure and weight gain. She reported episodes of dry mouth, intensive thirst and nocturia. Her blood pressure was 155/105 mm Hg, her skin was moderately atrophic and she could not perform squats, consistent with proximal muscle weakness. Her clinical signs of hypercortisolism were typical but discrete. She had no family history of adrenal tumors or arterial hypertension.
Her laboratory data showed normal serum sodium (143 mmol l−1), decreased serum potassium (2.8 mmol l−1), renin, ACTH and DHEAS, along with excessive serum levels of cortisol and aldosterone that could not be suppressed with fludrocortisone or medium-dose dexamethasone, respectively. Details of the endocrine function tests are given in Tables 2 and 3. After pretreatment with 25 mg per day of spironolactone for 6 weeks, retroperitoneoscopic adrenalectomy of the right adrenal gland was performed. Afterwards, the patient was put on hydrocortisone therapy that was gradually reduced until it could be ceased 9 months after surgery.
Methods
Clinical studies were performed according to a Clinical Practice Guideline.30 All tests were performed under this directive in an outpatient setting. Fludrocortisone at a dose of 0.1 mg was given orally four times a day for 4 days in combination with 40 mmol potassium three times a day and a salt-enriched diet. After a washout period of 2 weeks, dexamethasone was given at a dose of 0.5 mg four times a day for 3 days.
Immunohistochemical staining
For histological examination of the paraffin-embedded tumorous tissues (APA, CPA and A/CPA) and normal human adrenals, we used antibodies against the angiotensin II type 1 receptor (AT1R) (sc-81671, Santa Cruz Biotechnology, Santa Cruz, CA, USA; final dilution 1:200), ACTH receptor (MC2R) (sc-13107, Santa Cruz Biotechnology; final dilution 1:500) and 17α-hydroxylase (sc-46084, Santa Cruz Biotechnology; final dilution 1:500). For stainings, we used the catalyzed signal amplification system in combination with the biotin-blocking system (DAKO, Hamburg, Germany) and 3-amino-9-ethylcarbazole (AEC) as substrate chromogen (DAKO). The procedure was carried out according to the manufacturer's instructions and as described previously.31 All tissues were incubated for 30 min at room temperature. For control, material from APAs, CPAs and normal adrenal glands (NAG) was used.
Hormone measurements
Aldosterone, ACTH, DHEA-S and cortisol were measured by in-house routine immunoassays from patients’ plasma. Briefly, ACTH and cortisol were measured by an electrochemiluminescence -immunoassay (Elecsys, Roche, Mannheim, Germany). DHEA-S was measured by chemiluminescence immunoassay (Immulite, Siemens, Bad Nauheim, Germany). Aldosterone was measured by radio-immunoassay (Coat-a-Count, Siemens). 18-OH-F, 18-hydroxycorticosterone (18-OH-B), aldosterone-18-glucuronide and tetrahydroaldosterone were determined by the Steroid Laboratory of the Department of Pharmacology, University of Heidelberg, by radio-immunoassay as described elsewhere.32
Southern blot hybridization
For exclusion of a possible chimeric CYP11B1/CYP11B2 gene, Southern blotting was performed and followed by a long-range PCR, as described elsewhere.33
Western blot
Western blotting was performed to control the specificity of the antibody against 17α-hydroxylase and for confirmation of the immunohistochemical stainings in APA and CPA. According to the method of Laemmli, 40 μg protein were extracted from control tissue of APAs and CPAs and separated by electrophoresis in 10% SDS–PAGE. The adjacent transformation on an enhanced chemiluminescence (ECL) 0.45 μm nitrocellulose membrane was performed using the Bio-Rad (Munich, Germany) blotting system at 45 V for 105 min. After 1 h of blocking with 2 g BSA and 0.2 g skim milk powder in 40 ml TBS buffer, the membrane was incubated with the polyclonal anti-17α-hydroxylase antibody (sc-46084, Santa Cruz Biotechnology) at a 1:200 dilution at 4 °C overnight. As secondary antibody, a biotinylated polyclonal rabbit anti-goat immunoglobulin (E 0466, DAKO) was used at a dilution of 1:5000. Visualization was achieved by using the ECL-plus system (Amersham, Freiburg, Germany). For control, the membrane was incubated with primary monoclonal antibody against β-actin (Sigma-Aldrich, St Louis, MO, USA).
Results
Patients 1 and 2 exhibited typical clinical and biochemical features of both Cushing's and Conn's syndromes. Urinary excretion of tetrahydroaldosterone was 60 μg per day in patient 1 and 210 μg per day in patient 2 after the fludrocortisone-suppression test and a salt-enriched diet. The results of the endocrine function tests are given in Tables 2 and 3. After surgery on the adrenal tumors, the hormone-excess syndromes resolved. Both patients were negative for the hybrid CYP11B1/CYP11B2 gene.
Immunohistochemistry
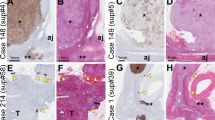
The tumors of both patients mainly comprised cells of a zona glomerulosa type. Remnant adrenal tissue was thin and histologically normal in both cases. We found a strong expression of 17α-hydroxylase in the patients’ tumors, in NAG, APA and CPA (Figure 1). AT1R protein expression was low in the patients’ tumors and CPA. Higher protein levels of AT1R were found in APA and NAG (Figure 1). Low levels of MC2R were seen in the tumors and CPA as compared to APA and NAG (Figure 1). As expected, normal adrenal medullary cells, which served as an internal negative control, did not react with the antibodies to 17α-hydroxylase, AT1R or MC2R.

Immunohistochemical analysis of corticotropin receptor (MC2R), angiotensin II type 1 receptor (AT1R) and 17α-hydroxylase (CYP17) expression in the patients’ adenomas (a–f) compared to aldosterone- (APA; g–i) and cortisol-producing adenomas (CPA; j–l) and normal human adrenal gland tissue (NAG; m–p). A positive immunoreaction is indicated by red staining, except for brown in the normal human adrenocortex stained for the ACTH receptor (lower left panel). The control inset represents a section of normal human adrenal cortex that was treated as the other tissue sections but without adding primary antibody.
Expression of 17α-hydroxylase
Western blotting of APA and CPA tissues with the anti-17α-hydroxylase antibody showed the presence of 17α-hydroxylase (58 kDa) and β-actin (42 kDa) protein in both CPA and APA tissue (Figure 2).

Western blot with antibodies to 17α-hydroxylase and β-actin proteins. Pure cortisol-producing adenomas (CPA) showed a typical band for 17α-hydroxylase (right lane). Pure aldosterone-producing adenomas (APA) also expressed the 17α -hydroxylase protein.
Discussion
PA is common among patients with treatment-resistant hypertension or hypertensive crises.30 Beginning with the first description in 1979,7, 8 an increasing number of cases with hypercortisolism and PA have been recognized in the recent years,9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 mainly in Japan. The majority of the reported cases have had subclinical autonomous glucocorticoid hypersecretion, whereas overt Cushing's syndrome has been found in only six patients. The true prevalence of A/CPAs may also be underestimated because hypercortisolism is not routinely excluded in patients with PA at most institutions. However, the removal of A/CPAs is reported to result in adrenal crisis,19 and our two patients depended on hydrocortisone replacement therapy after the operation. In addition, cortisol co-secretion by APA may also lead to the misinterpretation of hormonal ratios obtained during adrenal venous sampling because correcting the aldosterone values for autonomously secreted cortisol levels may give false-negative aldosterone-to-cortisol ratios at the side of the adenoma and a falsely low cortisol level in the contralateral vein. This can result in a low selectivity index.
Therefore, we suggest that a dexamethasone-suppression test be performed on each patient with PA before surgery or selective adrenal venous sampling. This may be of particular importance for patients with PA because of the comparatively large adrenal adenomas because A/CPAs tend to be larger than pure APAs,19, 20, 26 a tendency that held true for our patients.
Stowasser and Gordon differentiated between angiotensin II (AngII)-responsive and AngII-unresponsive APAs,34, 35, 36 which can be distinguished by the production of cortisol, cell type, extent of steroid suppression with dexamethasone and secretion of hybrid steroids, including 18-OH-F. Therefore, mildly elevated levels of 18-OH-F can be found in ordinary APAs.30 In addition, it has been shown as well that secreted cortisol can be metabolized to 18-OH-F by zona glomerulosa cells in normal subjects. Therefore, moderately elevated levels of hybrid steroids may just be due to hypersecretion of cortisol or aldosterone and its precursor 18-OH-B. In our patients, however, we found grossly elevated levels of 18-OH-F.
Elevated levels of 18-OH-F are also a typical finding in patients with familial hyperaldosteronism type 1 (FHA-I). FHA-I is a rare form of PA and may be diagnosed in patients with a family history of hypertension.37, 38 FHA-I is caused by an unequal cross-over between the CYP11B1 (11β-hydroxylase) and CYP11B2 (aldosterone synthase) genes.39, 40 This results in a chimeric CYP11B1/CYP11B2 gene, which puts aldosterone synthase activity under the control of corticotropin. As a result, hyperplastic adrenals of individuals with this chimeric gene can produce 18-OH-F from 18-OH-B.41 Notably, very high levels of 18-OH-F have also been found in FHA type 3 (FHA-III).42 The underlying genetic defect for this form of PA has not been identified. Yet, the family histories of our patients were negative, and a CYP11B1/CYP11B2 hybrid gene could not be identified.
Aldosterone is secreted by the adrenal glands in response to multiple stimuli (reviewed in Willenberg et al.43). Our two patients had undetectably low basal plasma levels of ACTH. The binding of ACTH to its receptor results in increased intracellular levels of cyclic AMP and expression of steroidogenic factor-1, both of which positively regulate MC2R.44, 45 Therefore, the low ACTH plasma concentrations in the two patients may explain the low levels of MC2R protein in both adenomas. Interestingly, the loss of sensitivity of adrenocortical cells from CPAs to their tropic stimulus ACTH has also been demonstrated in vitro,46 whereas in APAs, decreased AT1R expression and high MC2R expression was observed by us and also by others.47, 48, 49, 50
The levels of 17α-hydroxylase protein were high in both patients, pointing to a zona fasciculata cell type. The expression of 17α-hydroxylase in the tumors of patients with A/CPA has also been noted by others.20, 26 In addition, 17α-hydroxylase activity is reported to be similar in APA tissues as compared to normal human adrenal cortical tissue.51 This is in line with our experimental data showing that 17α-hydroxylase protein immunoreactivity was nearly as high in patients with A/CPA as in APAs, cortisol-producing adenomas and normal adrenal cortex. In a study that analyzed RNA expression levels, however, a higher ratio of CYP17/CYP11B2 messenger RNA was found in the patient's A/CPA tumor and in CPAs compared to pure APAs.26 Unfortunately, we were not able to perform expression studies at the RNA level.
The emergence of an APA from pure zona glomerulosa cells has been reported on rare occasions only.52 Therefore, immunohistochemistry for 17α-hydroxylase expression will most likely not result in specific detection of an A/CPA subtype of PA, as suggested by others.20 Instead, we suggest that laboratory criteria be applied for diagnosing A/CPA. The presence of an A/CPA should thus be considered if a patient exhibits both PA, substantiated by confirmatory testing6, 30 and non-suppressible cortisol in the dexamethasone-suppression test, elevated urinary free cortisol secretion or suppressed levels of corticotropin.53
In conclusion, these two cases lend support to the view that A/CPA constitutes a separate subtype of PA, with specific characteristics to be heeded during diagnostic and therapeutic procedures.
References
Gordon RD, Ziesak MD, Tunny TJ, Stowasser M, Klemm SA . Evidence that primary aldosteronism may not be uncommon: 12% incidence among antihypertensive drug trial volunteers. Clin Exp Pharmacol Physiol 1993; 20: 296–298.
Fardella C, Mosso L, Gomez-Sanchez C, Cortés P, Soto J, Gómez L, Pinto M, Huete A, Oestreicher E, Foradori A, Montero J . Primary hyperaldosteronism in essential hypertensives: prevalence, biochemical profile, and molecular biology. J Clin Endocrinol Metab 2000; 85: 1863–1867.
Mulatero P, Stowasser M, Loh KC, Fardella CE, Gordon RD, Mosso L, Gomez-Sanchez CE, Veglio F, Young Jr WF . Increased diagnosis of primary aldosteronism, including surgically correctable forms, in centers from five continents. J Clin Endocrinol Metab 2004; 89: 1045–1050.
Rossi GP, Bernini G, Caliumi C, Desideri G, Fabris B, Ferri C, Ganzaroli C, Giacchetti G, Letizia C, Maccario M, Mallamaci F, Mannelli M, Mattarello MJ, Moretti A, Palumbo G, Parenti G, Porteri E, Semplicini A, Rizzoni D, Rossi E, Boscaro M, Pessina AC, Mantero F, PAPY Study Investigators. A prospective study of the prevalence of primary aldosteronism in 1125 hypertensive patients. J Am Coll Cardiol 2006; 48: 2293–2300.
Rayner B . Primary aldosteronism and aldosterone-associated hypertension. J Clin Pathol 2008; 61: 825–831.
Stowasser M . Update in primary aldosteronism. J Clin Endocrinol Metab 2009; 94: 3623–3630.
Komiya I, Koizumi Y, Kobayashi R, Kotani M, Yamada T, Maruyama Y . Concurrent hypersecretion of aldosterone and cortisol from the adrenal cortical adenoma. Am J Med 1979; 67: 516–518.
Guthrie GP, Kotchen TA . Hypertension and aldosterone overproduction without renin suppression in Cushing's syndrome from an adrenal adenoma. Am J Med 1979; 67: 524–528.
Tunny TJ, Klemm SA, Gordon RD . Some aldosterone-producing adrenal tumours also secrete cortisol, but present clinically as primary aldosteronism. Clin Exp Pharmacol Physiol 1990; 17: 167–171.
Hobma S, Hermus A, Smals A, Kloppenborg P . Concurrent hypercortisolism and hyperaldosteronism due to an adrenal adenoma. Klin Wochenschr 1990; 68: 981–983.
Nagae A, Murakami E, Hiwada K, Kubota O, Takada Y, Ohmori T . Primary aldosteronism with cortisol overproduction from bilateral multiple adrenal adenomas. Jpn J Med 1991; 30: 26–31.
Imai T, Seo H, Murata Y, Funahashi H, Satoh Y, Sasano H, Matsui N, Takagi H . Dexamethasone-nonsuppressible cortisol in two cases with aldosterone-producing adenoma. J Clin Endocrinol Metab 1991; 72: 575–581.
Baert D, Nobels F, Van Crombrugge P . Combined Conn's and Cushing's syndrome: an unusual presentation of adrenal adenoma. Acta Clin Belg 1995; 50: 310–313.
Allan CA, Kaltsas G, Perry L, Lowe DG, Reznek R, Carmichael D, Monson JP . Concurrent secretion of aldosterone and cortisol from an adrenal adenoma—value of MRI in diagnosis. Clin Endocrinol 2000; 53: 749–753.
Makino S, Oda S, Saka T, Yasukawa M, Komatsu F, Sasano H . A case of aldosterone-producing adrenocortical adenoma associated with preclinical Cushing's syndrome and hypersecretion of parathyroid hormone. Endocr J 2001; 48: 103–111.
Honda T, Nakamura T, Saito Y, Ohyama Y, Sumino H, Kurabayashi M . Combined primary aldosteronism and preclinical Cushing's syndrome: an unusual case presentation of adrenal adenoma. Hypertens Res 2001; 24: 723–726.
Yamakita N, Murai T, Miyamoto K, Matsunami H, Ikeda T, Sasano H, Mune T, Yasuda K . Variant of pre-clinical Cushing's syndrome: hypertension and hypokalemia associated with normoreninemic normoaldosteronism. Hypertens Res 2002; 25: 623–630.
Tanaka M, Izeki M, Miyazaki Y, Horigome M, Yoneda T, Tsuyuki S, Takami S, Aiba M . Combined primary aldosteronism and Cushing's syndrome due to a single adrenocortical adenoma complicated by Hashimoto's thyroiditis. Intern Med 2002; 41: 967–971.
Sugawara A, Takeuchi K, Suzuki T, Itoi K, Sasano H, Ito S . A case of aldosterone-producing adrenocortical adenoma associated with a probable post-operative adrenal crisis: histopathological analyses of the adrenal gland. Hypertens Res 2003; 26: 663–668.
Adachi J, Hirai Y, Terui K, Nakano T, Fukuda Y, Suda T, Sasano H . A report of 7 cases of adrenal tumors secreting both cortisol and aldosterone. Intern Med 2003; 42: 714–718.
Markou A, Tsigou K, Papadogias D, Kossyvakis K, Vamvakidis K, Kounadi T, Piaditis G . A unique case of a benign adrenocortical tumor with triple secretion of cortisol, androgens, and aldosterone: development of multiple sclerosis after surgical removal of the tumor. Hormones 2005; 4: 226–230.
Fujii H, Kamide K, Miyake O, Abe T, Nagai M, Nakahama H, Horio T, Takiuchi S, Okuyama A, Yutani C, Kawano Y . Primary aldosteronism combined with preclinical Cushing's syndrome in an elderly patient. Circ J 2005; 69: 1425–1427.
Okura T, Miyoshi K, Watanabe S, Kurata M, Irita J, Manabe S, Fukuoka T, Higaki J, Sasano H . Coexistence of three distinct adrenal tumors in the same adrenal gland in a patient with primary aldosteronism and preclinical Cushing's syndrome. Clin Exp Nephrol 2006; 10: 127–130.
Sasaki N, Iwase M, Arima H, Nohara S, Bandai S, Yao T, Fujii K, Iida M . Overt diabetes mellitus in a patient with combined primary aldosteronism and Cushing's syndrome. Intern Med 2006; 45: 1237–1242.
Saito T, Ikoma A, Saito T, Tamemoto H, Suminaga Y, Yamada S, Kawakami M, Suzuki T, Sasano H, Ishikawa SE . Possibly simultaneous primary aldosteronism and preclinical Cushing's syndrome in a patient with double adenomas of right adrenal gland. Endocr J 2007; 54: 287–293.
Suzuki J, Otsuka F, Inagaki K, Otani H, Miyoshi T, Terasaka T, Ogura T, Omori M, Nasu Y, Makino H . Primary aldosteronism caused by a unilateral adrenal adenoma accompanied by autonomous cortisol secretion. Hypertens Res 2007; 30: 367–373.
Tsunoda K, Abe K, Yamada M, Kato T, Yaoita H, Taguma Y, Goto Y, Ioridani N . A case of primary aldosteronism associated with renal artery stenosis and preclinical Cushing's syndrome. Hypertens Res 2008; 31: 1669–1675.
Oki K, Yamane K, Sakashita Y, Kamei N, Watanabe H, Toyota N, Shigeta M, Sasano H, Kohno N . Primary aldosteronism and hypercortisolism due to bilateral functioning adrenocortical adenomas. Clin Exp Nephrol 2008; 12: 382–387.
Rossi E, Foroni M, Regolisti G, Perazzoli F, Negro A, Santi R, Grasselli C, Galli P, Gardini G . Combined Conn's syndrome and subclinical hypercortisolism from an adrenal adenoma associated with homolateral renal carcinoma. Am J Hypertens 2008; 21: 1269–1272.
Funder JW, Carey RM, Fardella C, Gomez-Sanchez CE, Mantero F, Stowasser M, Young Jr WF, Montori VM . Case detection, diagnosis, and treatment of patients with primary aldosteronism: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2008; 93: 3266–3281.
Haase M, Ansurudeen I, Paramonova I, Schinner S, Schott M, Papewalis C, Scherbaum WA, Willenberg HS . Evidence for the involvement of endothelial cell products in adrenal CITED2 expression. Cell Tissue Res 2009; 336: 337–343.
Vecsei P, Abdelhamid S, Mittelstadt GV, Lichtwald K, Haack D, Lewicka S . Aldosterone metabolites and possible aldosterone precursors in hypertension. J Steroid Biochem 1983; 19: 345–351.
Yokota K, Ogura T, Kishida M, Suzuki J, Otsuka F, Mimura Y, Oishi T, Hirata M, Tobe K, Makino H . Japanese family with glucocorticoid-remediable aldosteronism diagnosed by long-polymerase chain reaction. Hypertens Res 2001; 24: 589–594.
Stowasser M, Tunny TJ, Klemm SA, Gordon RD . Cortisol production by aldosterone-producing adenomas in vitro. Clin Exp Pharmacol Physiol 1993; 20: 292–295.
Gordon RD . Primary aldosteronism. J Endocrinol Invest 1995; 18: 495–511.
Stowasser M, Bachmann AW, Tunny TJ, Gordon RD . Production of 18-oxo-cortisol in subtypes of primary aldosteronism. Clin Exp Pharmacol Physiol 1996; 23: 591–593.
Sutherland DJ, Ruse JL, Laidlaw JC . Hypertension, increased aldosterone secretion and low plasma renin activity relieved by dexamethasone. Can Med Assoc J 1966; 95: 1109–1119.
New MI, Peterson RE . A new form of congenital adrenal hyperplasia. J Clin Endocrinol Metab 1967; 27: 300–305.
Lifton RP, Dluhy RG, Powers M, Rich GM, Cook S, Ulick S, Lalouel JM . A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature 1992; 355: 262–265.
Pascoe L, Curnow KM, Slutsker L, Conello JMC, Speiser PW, New MI, White PC . Glucocorticoid-suppressible hyperaldosteronism results from hybrid genes created by unequal crossovers between CYP11B1 and CYP11B2. Proc Natl Acad Sci USA 1992; 89: 8327–8331.
Mosso L, Gomez-Sanchez CE, Foecking MF, Fardella C . Serum 18-hydroxycortisol in primary aldosteronism, hypertension, and normotensives. Hypertension 2001; 38: 688–691.
Geller DS, Zhang J, Wisgerhof MV, Shackleton C, Kashgarian M, Lifton RP . A novel form of human mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab 2008; 93: 3117–3123.
Willenberg HS, Schinner S, Ansurudeen I . New mechanisms to control aldosterone synthesis. Horm Metab Res 2008; 40: 435–441.
Naville D, Penhoat A, Durand P, Begeot M . Three steroidogenic factor-1 binding elements are required for constitutive and cAMP-regulated expression of the human adrenocorticotropin receptor gene. Biochem Biophys Res Commun 1999; 255: 28–33.
Sarkar D, Kambe F, Hayashi Y, Ohmori S, Funahashi H, Seo H . Involvement of AP-1 and steroidogenic factor (SF)-1 in the cAMP-dependent induction of human adrenocorticotropic hormone receptor (ACTHR) promoter. Endocr J 2000; 47: 63–75.
Willenberg HS, Stratakis CA, Marx C, Ehrhart-Bornstein M, Chrousos GP, Bornstein SR . Aberrant interleukin-1 receptors in a cortisol-secreting adrenal adenoma causing Cushing's syndrome. N Engl J Med 1998; 339: 27–31.
Ganguly A . Cellular origin of aldosteronomas. Clin Investig 1992; 70: 392–395.
Schubert B, Fassnacht M, Beuschlein F, Zenkert S, Allolio B, Reincke M . Angiotensin II type 1 receptor and ACTH receptor expression in human adrenocortical neoplasms. Clin Endocrinol 2001; 54: 627–632.
Oda N, Takeda Y, Zhu A, Usukura M, Yoneda T, Takata H, Mabuchi HI . Pathophysiological roles of the adrenal renin-angiotensin system in patients with primary aldosteronism. Hypertens Res 2006; 29: 9–14.
Mazzuco TL, Lampron A, Bourdeau I, Lacroix A . Aberrant hormone receptors in primary aldosteronism. Horm Metab Res 2010 (e-pub ahead of print; in press).
Suzuki H, Shibata H, Maruyama T, Ishimura Y, Saruta T . Significance of steroidogenic enzymes in the pathogenesis of hyperfunctioning and non-hyperfunctioning adrenal tumor. Steroids 1995; 60: 42–47.
Shigematsu K, Nishida N, Sakai H, Igawa T, Suzuki S, Kawai K, Takahara O . Primary aldosteronism with aldosterone-producing adenoma consisting of pure zona glomerulosa-type cells in a pregnant woman. Endocr Pathol 2009; 20: 66–72.
Nieman LK, Biller BMK, Findling JW, Newell-Price J, Savage MO, Stewart PM, Montori VM . The diagnosis of cushing's syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol 2008; 93: 1526–1540.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Willenberg, H., Späth, M., Maser-Gluth, C. et al. Sporadic solitary aldosterone- and cortisol-co-secreting adenomas: endocrine, histological and genetic findings in a subtype of primary aldosteronism. Hypertens Res 33, 467–472 (2010). https://doi.org/10.1038/hr.2010.18
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2010.18
Keywords
This article is cited by
-
A patient with concurrent primary aldosteronism and Page kidney
Endocrine (2010)
