
Abstract
Since their inception, angiotensin-converting enzyme (ACE) inhibitors have been used as first-line therapy for the treatment of cardiovascular and renal diseases. They restore the balance between the vasoconstrictive salt-retentive and hypertrophy-causing peptide angiotensin II (Ang II) and bradykinin, a vasodilatory and natriuretic peptide. As ACE is a promiscuous enzyme, ACE inhibitors alter the metabolism of a number of other vasoactive substances. ACE inhibitors decrease systemic vascular resistance without increasing heart rate and promote natriuresis. They have been proven effective in the treatment of hypertension, and reduce mortality in congestive heart failure and left ventricular dysfunction after myocardial infarction. They inhibit ischemic events and stabilize plaques. Furthermore, they delay the progression of diabetic nephropathy and neuropathy and act as antioxidants. Ongoing studies have elucidated protective roles for them in both memory-related disorders and cancer. Lastly, N- and C-domain selective ACE inhibitors have led to new uses for ACE inhibitors.
Similar content being viewed by others
Introduction
In recent years, stressful and fiercely competitive lifestyles and food habits have compounded the problems of hypertension. Long-standing and stressful, progressively rising hypertension can lead to many disorders, including myocardial infarction (MI), cerebrovascular events, congestive heart failure, peripheral arterial insufficiency, premature mortality1 and renal dysfunction leading to glomerulosclerosis and kidney artery aneurysm.2
A number of therapies are available, but angiotensin-converting enzyme (ACE) inhibitors have been the preferred first-line therapy for hypertension, congestive heart failure, left ventricular (LV) systolic dysfunction and MI.3, 4 ACE inhibitors (ACEis) have been in use for the past two decades, and the interest in them is still growing. Recently, the discovery of domain-selective ACEis and new members of the renin–angiotensin system (RAS) (that is, angiotensin-converting enzyme 2) have again fueled the interest of researchers. Some new studies have expanded the already impressive clinical profile of ACEis. This review traces some already known and new facets of ACE inhibition and introduces new advances in the designing of a new generation of ACEis.
Angiotensin-converting enzyme inhibitors
The ACE (or kininase II) is a bivalent dipeptidyl carboxyl metallopeptidase, which is a membrane enzyme in endothelial, epithelial and neuroepithelial cells and the brain; it is also present in a soluble form in blood and numerous body fluids.5 ACE cleaves the C-terminal dipeptide from Ang I and bradykinin, thus interacting with RAS and the kallikrein–kinin system simultaneously.
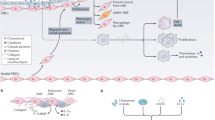
It can be concluded that ACE has a pivotal role in the balance between vasodilatory and natriuretic properties of bradykinin, and vasoconstrictive and salt-retentive properties of Ang II. An increase in ACE activity disturbs this delicate balance and promotes vasoconstrictive and salt-retentive Ang II (Figure 1) and decreases vasodilatory and natriuretic bradykinin. The ACEis restore this balance by decreasing the formation of Ang II and the degradation of bradykinin (Figure 2).

Classical view of the action of angiotensin-converting enzyme (ACE) on angiotensin (Ang) I and the effect of Ang II on different tissues.

Effect of angiotensin-converting enzyme (ACE) inhibition on blood pressure and independent actions of angiotensin (Ang II).
A number of ACEis are currently in the market. The ACEis differ in the chemical structure of their active moieties, as well as in their potency, bioavailability, plasma half-life, route of elimination, distribution and affinity for tissue-bound ACE, and whether they are administered as prodrugs. The ACEis may be classified into three groups according to the chemical structure of their active moiety. Captopril is the prototype of the sulfhydryl-containing ACEis; others are fentiapril, pivalopril, zofenopril and alacepril. Fosinopril is the only ACE inhibitor containing a phosphinyl group as its reactive moiety. The majority of other ACEis, like lisinopril, enalapril and perindopril, contain a carboxyl moiety.6
Clinical implications of ACE inhibition
Essential hypertension
According to The Joint National Committee (JNC) VII, ACEis are one of the first-line drugs for hypertension. Patients with stage-I hypertension (systolic blood pressure (BP): 140–159 or diastolic BP: 90–99 mmHg) should be treated with ACEis.7 The Antihypertensive and Lipid Lowering Therapy in Heart Attack Trial (ALLHAT) also suggests the use of ACEis as initial therapy alone or in combination with thiazide diuretics, with an overall response of 50–70% in mild to moderate disease.8
ACEis are effective in lowering the mean, systolic and diastolic pressures in hypertensive patients as well as in salt-depleted normotensive subjects.9, 10, 11 The acute change in BP correlates with pretreatment plasma renin activity and angiotensin levels, such that the greatest reductions in BP are seen in patients with the highest plasma renin activity.12, 13, 14, 15 In support of this, co-administration of drugs that increase plasma renin activity, such as diuretics, abolishes the racial differences in response to ACEis.16 However, with long-term therapy, a greater percentage of patients achieve a decrease in BP, and the antihypertensive effect no longer correlates with pretreatment plasma renin activity.13, 14, 15 The mechanism for this increased efficacy with chronic administration is not clear but may involve the kallikrein–kinin system or production of vasodilatory prostaglandins.16
One of the characteristics of ACEis is that they lower peripheral vascular resistance without causing a compensatory increase in heart rate17, 18, 19, 20or changing baroreceptor activity.21 They also show lack of reflex tachycardia22 and cause inhibition of the normal tonic influence of Ang II on the sympathetic nervous system.23 During ACE inhibition, heart rate responses to postural changes and exercise are not impaired.24
Atherosclerosis
In a recent study, it was observed that the antiatherosclerotic effect of quinapril is more potent than that of losartan in hypertensive patients, as quinapril reduced carotid artery intima-media thickness by a larger extent in patients with mild-to-moderate arterial hypertension than did losartan.25
Experimental evidence shows that ACE inhibition can retard the development of atherosclerosis. In animal models for atherosclerosis, including apolipoprotein E knockout mice,26 Watanabe rabbits27 and cholesterol-fed monkeys,28 the ACEis reduce the extent of vascular lesions. Oxidatively modified low-density lipoprotein (LDL) has an important role in the development of atherosclerosis. ACE inhibition retards the development of atherosclerosis by reducing expression of LOX-1, the human endothelial receptor for oxidized LDL, in mammary artery biopsies of patients with coronary artery disease.29 Furthermore, endothelial dysfunction in normotensive patients with coronary artery disease, hypertensive patients and patients with non-insulin-dependent diabetes mellitus is reversed by ACE inhibition.30 The ACEis also improve endothelial function by attenuating the vasoconstrictive and superoxide radical-generating effects of Ang II while simultaneously enhancing the bradykinin-dependent induction of endothelial nitric oxide (NO) production. The ACEis promote ischemic preconditioning, probably through a bradykinin-mediated mechanism.31 Studies in vitro and in humans suggest that ACEis favorably alter the fibrinolytic balance, both by decreasing Ang II, a potent stimulus for plasminogen activator inhibitor synthesis,32, 33 and by increasing bradykinin, a potent stimulus for tissue plasminogen activator secretion.34, 35 Trials like Heart Outcomes Prevention Evaluation (HOPE) suggest that ramipril, an ACE inhibitor, is beneficial in a broad range of patients with no evidence of LV systolic dysfunction or heart failure who are at high risk for cardiovascular events. Treatment with ramipril reduced the rates of death, MI, stroke, coronary revascularization, cardiac arrest and heart failure, as well as the risk of complications related to diabetes and diabetes itself.36 In addition to decreasing Ang II and increasing bradykinin, a study demonstrated that the antiatherogenic effects of ACEis are also dependent on their active groups. Chronic treatment with sulfhydryl ACEis, like zofenopril and captopril, is more successful at reducing the susceptibility of plasma LDL to in vitro oxidation, formation of oxidation-specific epitopes in the arterial wall and atherogenesis in apolipoprotein E knockout mice.37
Congestive heart failure and LV dysfunction
Since 1987, several large, prospective, randomized, placebo-controlled trials have demonstrated that treatment with ACEis results in a reduction in overall mortality in patients with congestive heart failure due to systolic dysfunction.38, 39, 40, 41, 42 The reduction in mortality has even been seen in patients with asymptomatic LV dysfunction.43, 44 This reduction in mortality results primarily from a reduction in the progression of congestive heart failure,38, 40, 41 although the incidence of sudden death40 and MI43may also decrease.
Although ACEis improve outcome in patients with systolic dysfunction, many patients with hypertension experience congestive heart failure due to diastolic dysfunction related to LV hypertrophy. In animal models, ACEis have been shown to reverse ventricular remodeling by blocking the trophic effects of Ang II on cardiac myocytes.45, 46 The ACEis have been shown to reverse LV hypertrophy in patients with hypertension.19, 47, 48 A meta-analysis of the effects of several antihypertensive agents suggested that ACEis were the most effective agent for reducing LV hypertrophy.49
Myocardial infarction
Animal models and human studies demonstrated that ACEis attenuate LV remodeling after MI. Subsequently, large, randomized, controlled trials have shown that ACEis reduce cardiovascular and all-cause mortality, prevent or delay the onset of heart failure, and decrease the risk of stroke after a MI.50 Early ACE inhibitor treatment (⩽36 h post-MI) is associated with significantly lower mortality both in the first week and in the first month after MI.51 Long-term trials such as the Survival and Ventricular Enlargement (SAVE) trial, the Acute Infarction Ramipril Efficacy (AIRE) study and the Trandolapril Cardiac Evaluation (TRACE) study selectively enrolled high-risk patients with LV dysfunction or heart failure after MI and demonstrated a 20% reduction in mortality.52 ACEis prevent or delay the detrimental cardiac remodeling after MI that apparently contributes to the worse prognosis after MI as shown by perindopril in a recent Perindopril and Remodeling in Elderly with Acute Myocardial Infarction (PREAMI) trial.53 According to class I American College of Cardiology/American Heart Association guidelines,54 The ACEis should be given to all patients with acute MI and anterior ST elevation, an ejection fraction of 40% or clinical heart failure in the absence of hypotension. Therapy should be continued for at least 6 weeks in all patients and indefinitely in patients with persistent LV dysfunction, although another study suggests a role for long-term ACE inhibition in all patients with ischemic heart disease.55
Diabetic nephropathy
Both RAS and increased glomerular capillary pressure have been implicated in the progression of renal dysfunction due to a number of renal diseases, including diabetic nephropathy.56 In addition, Ang II causes mesangial cell growth and matrix production.57, 58 The ACEis reduce glomerular capillary pressure by decreasing arterial pressure and selectively dilating efferent arterioles.59 Numerous animal studies and small clinical trials have suggested that ACEis significantly reduce the loss of kidney function in diabetic nephropathy.60 ACEis prevent the progression of microalbuminuria to overt proteinuria.61 A large, prospective, placebo-controlled study has shown that captopril slows the progression of nephropathy in patients with insulin-dependent diabetes mellitus, as measured by the rate of decline in creatinine clearance and the combined end points of dialysis, transplantation and death.62 A second large-scale, prospective, double-blind study extended these observations by showing a protective effect of ACEis in patients with a variety of renal diseases, including glomerulopathies, interstitial nephritis, nephrosclerosis and diabetic nephropathy.63 The exception was polycystic kidney disease. Importantly, the protective effect of ACE inhibition was independent of the severity of renal insufficiency.
In a recent study, it was observed that blockade of the RAS by ACEis in type-2 diabetis patients with diabetic nephropathy reduces urinary monocyte chemoattractant protein-1 levels and improves renal function. As monocyte chemoattractant protein-1 induces monocyte immigration and differentiation to macrophages, which augment extracellular matrix production and tubulointerstitial fibrosis, pharmacological reduction of Ang II may also exert its beneficial effects in diabetic nephropathy by downregulation of renal monocyte chemoattractant protein-1.64
Diabetic neuropathy
Angiotensin II increases oxidative stress in the kidney by stimulating NAD(P)H oxidase,65, 66, 67 which is a primary source of reactive oxygen species in vascular tissue and a contributing factor in diabetic neuropathy.68, 69
In addition to nephropathy and hypertension, ACEis are also effective against diabetic neuropathy. In diabetic rats, lisinopril treatment prevented nerve dysfunction.70 In studies of human diabetes, two small clinical trials demonstrated that diabetic neuropathy was improved by treatment of patients with trandolapril or lisinopril.71, 72
Diabetes causes impairment in vascular relaxation in response to acetylcholine and calcitonin gene-related peptide in epineurial arterioles of the sciatic nerve.73, 74 Acetylcholine-induced vascular relaxation is endothelium dependent and mediated by NO and endothelium-derived hyperpolarizing factor, and both are impaired by diabetes.73, 75, 76 Moreover, impairment by diabetes of acetylcholine-mediated vascular relaxation of epineurial arterioles precedes the slowing of motor nerve conduction velocity suggesting that vascular dysfunction contributes to impaired nerve activity.77 In a study by Coppey et al.,78 it was demonstrated that the ACE inhibitor enalapril was more effective than the angiotensin 1 (AT1) receptor blocker, L-158809, at reversing diabetes-induced impairment of endoneurial blood flow and motor nerve conduction velocity. Furthermore, enalapril treatment was more capable of preventing/reversing the diabetes-induced impairment of relaxation by acetylcholine and calcitonin gene-related peptide. It was demonstrated that treatment of diabetic rats with enalapril reduced superoxide formation by 50–75% in epineurial arterioles of the sciatic nerve, and with L-158809 treatment the decrease in superoxide level in epineurial arterioles was minimal. The improvement of acetylcholine-mediated vascular relaxation may be due to increased formation of NO capable of overcoming quenching by superoxide and/or improved formation/activity of endothelium-derived hyperpolarizing factor.79, 80, 81 In this regard, Kihara et al.82 have demonstrated that treatment of diabetic rats with ACEis improves diabetic neuropathy by increasing NO synthase synthesis.
ACE inhibitors as antioxidant
In addition to reducing levels of Ang II and increasing bradykinin, ACEis have important implications for vascular oxidative stress. All major cell types of the vascular wall (endothelium, smooth muscle and fibroblasts) contain the enzyme NADPH oxidase, which is responsible for the production of superoxide anion,83 which is activated in response to Ang II84 and leads to both hypertension85 and smooth-muscle cell hypertrophy.86 Experimental models of atherosclerosis have also demonstrated activation of the local RAS and vascular NADH/NADPH oxidase activity.87
The ACEis represent a new antioxidant strategy that targets oxidative stress at its source. ACE inhibition, by decreasing Ang II, limits the stimulation of vascular NAD(P)H oxidase, thereby preventing the increased superoxide flux associated with activation of the RAS.88 As superoxide reacts with NO, ACE inhibition also improves NO bioactivity, as has been observed in patients with coronary artery disease89 and in experimental models for hypertension.90 The ACEis limit the formation of H2O2, which is responsible for smooth muscle cell proliferation, and this slows the progression of carotid intimal thickening as described in the study to evaluate carotid ultrasound changes in patients treated with ramipril and vitamin E (SECURE).91
The antioxidant effect of ACEis is BP independent. In vivo, ACE inhibition by zofenopril for 6 weeks in Watanabe heritable hyperlipidemic rabbits reduced plasmatic LDL oxidation and atherosclerosis.92 In vitro, a number of ACEis (thiols and nonthiols) showed a direct inhibitory effect on LDL oxidation in the presence of copper.93 Angiotensin II can increase 12-lipoxygenase activity in vascular smooth muscle cells94 and macrophage lipoxygenase activity through AT1 receptor (AT1R)-mediated mechanisms.95 Moreover, Ang II-mediated macrophage lipid peroxidation was found to involve the action of cellular NADPH oxidase as well as 15-lipoxygenase.96 It has been suggested that lipoxygenase and NADPH oxidase mediate the oxidative modification of LDL.97 Thus, the antioxidative effect of ACEis seems to be mediated by decreasing the expression of the lipoxygenase enzyme.
Plaque stabilization
Angiotensin II stimulates endothelin release. Endothelin is one of the most potent coronary vasoconstrictors, and its local release might, in the presence of a susceptible atherosclerotic lesion, accelerate plaque rupture.98, 99 Hypomagnesemia has also been shown to cause an increase in coronary vascular reactivity and could potentially accelerate plaque rupture.100, 101 Ang II can trigger most of the changes reported to be associated with magnesium deficiency, including induction of pro-oxidant and proinflammatory conditions.102 Moreover, Ang II induces the synthesis of plasminogen activator inhibitor type 1 in endothelial cells. Therefore, activated RAS accelerates the formation of coronary artery plaque and thrombus associated with hypertension. This results in destabilization of arteriosclerotic plaques.103
The ACEis have a role in reducing the propensity for plaque rupture. A reduction in BP and pulse rate with the use of ACEis may reduce the propensity for plaque disruption by reducing circumferential stress on the fibrous caps of lipid-rich plaques.104 Other mechanisms of preventing plaque rupture include reduced levels of neurohumoral activation and effects on protein synthesis influencing plaque composition as Ang II stimulates vascular smooth muscle growth and proliferation. The prevention of ischemic events by ACEis also involves magnesium. Barbagallo et al.105 showed that magnesium influences cellular response to ischemia and that the ability of thiol compounds, such as captopril, to ameliorate ischemic injury may be, at least in part, due to their ability to increase cytosolic free magnesium levels.
Memory-enhancing effect of ACEis
In 2002, Amenta et al.106 reviewed the majority of controlled clinical trials and observed that ACEi treatment (including perindopril, captopril and lisinopril) positively influenced cognitive function independently of its BP-lowering effects, and patients treated with ACEi displayed better results than those on diuretics and β-blockers. A number of animal studies on the cognitive effects of ACEis have also corroborated this observation. Treatment with 24 mg/kg/day enalapril in streptozotocin-treated rats for 14 weeks from the onset of diabetes significantly improved memory performance in the water maze compared to untreated diabetics.107 Long-term administration of the ACE inhibitor captopril (400 mg/l through the drinking water), either from the time of weaning or from the age of 6 months (that is, several months after hypertension was established), improved performance in working memory spatial task in both spontaneously hypertensive rats and WKY at 24 months of age.108
Even in clinical situations the use of ACEis has prevented cognitive decline in patients. The Perindopril Protection Against Recurrent Stroke Study (PROGRESS), a large, randomized, double-blind, placebo-controlled trial conducted among patients with prior stroke or transient ischemic attack, also showed that ACEi has positive effects on memory. The treatment group showed a risk reduction in cognitive decline of 19%, and the composite risks of dementia with recurrent stroke and of cognitive decline with recurrent stroke declined by 34 and 45%, respectively.109 These findings indicate that an increase in cerebral perfusion may be responsible for the observed reduction in cognitive deficit after ACEi treatment. The mechanism behind the positive effect of ACEis on memory is ambigous. The ACE is a promiscuous enzyme; apart from Ang II and bradykinin, a number of other neuropeptide substrates have been reported for ACE, including substance P, neurotensin, dynorphin and enkephalin.110 Some of these neuropeptide substrates have memory-enhancing properties; substance P, after peripheral or central application, has reinforcing and memory-facilitating effects inducing place preference111, 112 and avoidance learning;113, 114 dynorphins have an improving role in memory-impairment paradigms, such as basal forebrain lesions115 and mecamylamine-induced116 and galanin-induced impairment117 of learning and memory.
ACEi treatment has been shown to increase hippocampal blood flow and improve hippocampal long-term potentiation in diabetic rats compared with untreated diabetics.107 These results suggest that ACEi may act through the hippocampal acetylcholine system, which is essential for memory,118, 119 to exert their positive effects on cognition.
Anticancer effect of ACEis
The RAS120 and angiotensin receptors121 have such an important role in cancer that the Ang II–AT1R system has begun to be viewed as a therapeutic target for cancer.122, 123 Ang II has an anti-apoptotic role involving AT1R/phosphatidylinositol 3-kinase/Akt activation and the subsequent suppression of activation of caspase-9124 and caspase-3.125 Ang II also reduces cell adhesion and invasion through the type 1 receptor, and this effect may be due to reduced expression of integrins, in particular α3 and β1.126 Ang II acts through AT1Rs to enhance vascular endothelial growth factor expression resulting in endothelial cell proliferation, migration and angiogenesis.127, 128 Activation of AT1R is also reported to induce inflammatory cytokines and chemokines through mitogen-activated protein kinase, reactive oxygen species and nuclear factor-κB pathways129, 130 (Figure 2). All these processes mediated by AT1 are important in the induction and progression of cancer.
Several in vivo studies in tumor models have revealed that ACEis inhibit tumor growth131and angiogenesis.132, 133 The beneficial effect of ACEis in suppressing tumor growth is primarily mediated through RAS-dependent inhibition of Ang II levels and increases in bradykinin levels.134 Another anti-cancer activity of some ACEis is probably due to their intrinsic metal-chelating properties, which are thought to be responsible for inhibition of matrix metalloproteinase.135, 136 The direct effect of some ACEis is the reduction of plasminogen activator inhibitor and another effect is due to their free sulfhydryl group, which leads to generation of angiostatin, which in turn inhibits angiogenesis.137 The sulfhydryl group present in ACE inhibitor also serve as a free radical scavenger.138 The reduction of reactive oxygen species prevents subsequent activation of matrix metalloproteinase and vascular endothelial growth factor, thus preventing further invasion and angiogenesis as shown in Figure 2.
However, a few reports have suggested that ACEis may actually promote tumor growth, possibly due to their immunomodulatory activity,139 and patients treated with ACEis have been reported to have higher rates of kidney cancers.140 However, several other clinical studies have shown that ACEis do not increase the risk of cancer.141, 142 A prospective study in Glasgow in 5027 patients has shown that ACEis may protect against cancer143 and are effective in managing post-anthracycline cardiotoxicity in pediatric cancer survivors without increasing any risk of cancer.144 The role of ACE in cancer is further supported by the findings that ACE polymorphism has been found to be associated with several cancers, including prostate, gastric and breast cancer.145, 146, 147
Thus, several studies support anti-cancer effects of ACEis through RAS-dependent and/or -independent mechanisms, and these inhibitors may help us understand interactions between components of RAS in tumorigenesis and design new anti-cancer agents.
Local RAAS: target organ protection and ACEis
Renin–angiotensin–aldosterone system (RAAS) is also present in a wide variety of organs (tissue RAAS) promoting the local synthesis of Ang II. This local RAAS system has an important role in the development of hypertensive target organ damage.148 Apart from reducing BP, ACEis also obstruct local RAAS and protect against target organ damage, reversing or preventing its development
Experimental studies in rats demonstrated that treatment of spontaneously hypertensive rats for 12 weeks with either antihypertensive or non-antihypertensive ACEis reduced interstitial fibrosis and normalized ventricular stiffness, whereas normalization of BP and regression of cardiac hypertrophy were achieved only with an antihypertensive dosage of ACEi. They also confirmed that regression of fibrosis is independent of changes in BP and cardiac hypertrophy.149
The presence of local RAAS also extends to kidneys. Local activation of RAAS causes glomerular injury by further raising glomerular capillary pressure through Ang II-induced vasoconstriction of the efferent arterioles.150 RAAS may also cause proteinuria through an effect on the expression of renal nephrin, a transmembrane protein located in glomerular podocytes. ACEis decrease proteinuria and delay the progression of renal disease in patients with nondiabetic kidney disease. In the Angiotensin-Converting Enzyme Inhibition in Progressive Renal Insufficiency (AIPRI) study, benazepril caused a 53% reduction of the overall risk in terms of reducing serum creatinine and need for dialysis in nondiabetic patients with chronic kidney disease as compared with placebo. In this trial, the benefit of ACE inhibition was greatest in patients with the highest level of proteinuria.151
Endothelial dysfunction seems to be a marker of disease progression and of increased risk of cardiovascular morbidity and mortality.152 In a study by Ghiadoni et al.,153 treatment with nifedipine, amlodipine, atenolol, nebivolol, telmisartan and perindopril similarly reduced BP, but only perindopril increased flow-mediated endothelium-dependent dilation in conduit arteries without modifying the response to glyceryl trinitrate.
Looking beyond ACE inhibition
The ACE is a promiscuous enzyme; apart from Ang II and bradykinin, a number of other neuropeptide substrates have also been reported for ACE, including substance P, neurotensin, dynorphin, and enkephalin.110 For this reason ACEis have diverse effects independent of their BP-lowering effect (Figure 2). Indeed, different ACEis like moexiprilate, ramipirilate, captopril, enalpril and quinaprilate amplified the effects of bradykinin in vessels that lacked measurable ACE activity154 and also enhanced the effect of an ACE-resistant B2-kinin receptor agonist. These findings demonstrate that ACEis selectively potentiate the B2-receptor-mediated vascular effects of bradykinin independently of their ACE-inhibiting properties, and this might be related to differences in molecular structure.154, 155 ACEis also induce cross-talk between the transmembrane protein ACE and the B2-kinin receptor, probably by formation of a heterodimer. This protects high-affinity receptors, blocks receptor desensitization and decreases internalization, thereby potentiating BK beyond blocking its hydrolysis.156, 157 ACEis also directly activate the bradykinin B1 receptor by acting at the zinc-binding pentameric consensus sequence HEXXH (195–199) of the B1 receptor, a motif that is present in the active center of ACE but absent from the B2 receptor. ACEis, when activating the B1 receptor, elevate intracellular calcium ([Ca2+]i) and release NO from cultured cells.158
ACEis also induce phosphorylation of the ACE intracellular tail (Ser1270) via CK2, resulting in outside-in signaling that enhances expression of ACE and cyclooxygenase-2.159 The effect of the ACE inhibitor on cyclooxygenase-2 is due to the transcription factor activator protein-1. This results in increased release of prostacyclin and prostaglandin E2 by endothelial cells, which is independent of local accumulation of kinins.160
In addition to the kallikrein–kinin system, ACE also interacts with the opioidergic system. Hypertension and reduced pain perception have been repeatedly associated in animals and in humans. A study investigating pain threshold by hot-plate test in normotensive Wistar-Kyoto rats and spontaneously hypertensive rats after ACE inhibitor treatment observed a normalization of increased hot-plate latencies, whereas pain sensitivity was unaffected by hydralazine, which is otherwise effective at reducing BP.161 Hypertensive patients show a reduced sensitivity to pain that is independent of the method used to induce the painful stimulus.162, 163 Takai et al.163 investigated the involvement of endogenous brain Ang II in nociception in mice by using ACEis and an Ang II antagonist. Significantly longer jump latencies were obtained for the groups repeatedly treated with spirapril, trandolapril and losartan, and these antinociceptive effects were reversed by naloxone.
Clinical studies also confirm an interaction between the RAS and endogenous opioids. Guasti et al.,164 using dental pain perception evaluation by means of a pulpar test, showed that the pain threshold was significantly higher in hypertensive than in normotensive subjects. However, the pain threshold and tolerance in the patients decreased to levels similar to those of normotensives during treatment with enalapril. Kalra et al.165 evaluated the effect of ramipril (an ACEi) and losartan (an AT1R blocker (ARB)) on pain perception in human volunteers and observed that groups receiving either ramipril or losartan showed a decline in the threshold for maximum tolerated pain, but only ramipril (and not losartan) decreased the pain perception threshold.
Adverse effect of ACEis
Like all antihypertensive agents, ACEis can cause hypotension particularly in patients with elevated plasma renin activity. Therefore, lower starting doses should be used under these conditions.166 ACEis can cause hyperkalemia because of a decrease in aldosterone,167 especially in patients with impaired kidney function or in patients who are taking potassium supplements (including salt substitutes) or potassium-sparing diuretics.168, 169 ACEis can cause a reversible decline in renal function in the setting of decreased renal perfusion due to bilateral renal artery stenosis,170 severe congestive heart failure171 or volume depletion.172
Coughing is a frequent side effect of ACEis. The mechanism is not known but may involve increased levels of bradykinin or substance P and stimulation of vagal C fibers.173 Thromboxane antagonism, aspirin and iron supplementation reduce coughing induced by ACEis.166 Angioedema, a rare but potentially life-threatening side effect of ACEis, is characterized by localized swelling of the lips, tongue, mouth, throat, nose or other parts of the face. The mechanism seems to involve bradykinin or one of its metabolites.173
If administered in the second or third trimester of pregnancy, ACEis can cause oligohydramnios, fetal growth retardation, pulmonary hypoplasia, joint contractures, hypocalvaria, neonatal renal failure, hypotension and death. These effects result from blockade of the conversion of Ang I to Ang II in the developing fetal kidneys leading to fetal hypotension.174 For this reason, ACEis should be stopped once a pregnancy has been confirmed.
Adverse effects that seem to be related to the presence of a sulfhydryl group are neutropenia, nephrotic syndrome and skin rash.175 The incidence is higher in patients who have renal insufficiency or collagen vascular disease. Skin rashes occur in 1% of patients and usually consist of a pruritic maculopapular eruption; rarely, exfoliative dermatitis has been reported. The rash seems to be dose related.6
ACEis vs. AT1R blockers (ARBs)
Accumulating data show that ARBs are on a par with ACEis in a number of clinical benefits. However, it is now clear that ARBs and ACEis have some distinct differences in their beneficial effects as evidenced by clinical studies using ACEis and ARBs.
One factor, the effect on bradykinin, is likely to favor ACE inhibition over ARB therapy. As discussed above, ACE inhibition prevents the breakdown of bradykinin, which is a vasodilatory peptide. The effect of ACE inhibition became attenuated when bradykinin receptor antagonists were used, suggesting that bradykinin has an essential role in the antihypertensive action of ACEis.176
The Blood Pressure Lowering Treatment Trialist's Collaboration (BPLTT) showed comparable BP-dependent reductions in risk with ACEis and ARBs, but only ACEis elicited a statistically significant BP-independent reduction in relative risk of coronary disease events of 9%, whereas no such effect was reported with ARBs.177 Similarly, Strauss and Hall178 reported a consistent reduction in MI and cardiovascular death with ACEis, but a number of ARB trials in high-risk patients demonstrated an almost complete lack of reduction in MI and mortality despite significant reductions in BP. Nine of 11 key clinical ARB trials have even reported an excess of MI that achieved statistical significance in two cases: VALUE (VAlsartan antihypertensive Long-term Use Evaluation) and CHARM-Alternative.
However, in the case of stroke prevention, treatment with ARBs as compared with that using ACEis provides neuroprotection from focal cerebral ischemia in animal studies.179 In this neuroprotection by ARB, AT2 receptor has an important role. Stimulation of the AT2 receptors in neurons in the brain region adjacent to the infarct area induces vasodilatation by locally synthesized NO and prostacyclin, thus improving cerebral blood flow by collateral circulation.180, 181 Selective blockade of central AT2 receptors abolishes the neuroprotective effect of ARBs.180
A limitation of ACEis in preventing stroke is further exhibited in PROGRESS, which showed that the ACE inhibitor perindopril caused only a 5% reduction in stroke, compared with a 43% stroke reduction when the diuretic, indapamid, was added to the ACE inhibitor.109 This finding can be explained on the basis of the hypothesis of Fournier et al.,182 which states that in stroke prevention, diuretics, calcium antagonists and ARBs, which increase Ang II formation, have an edge over ACEis and β-blockers, which decrease Ang II formation. However, in the HOPE study, ramipril caused a 32% reduction of strokes because it reduced cardiac complications three-fold and prevented plaque destabilization, which otherwise could have caused stroke.182
To settle the debate regarding ACEis vs. ARBs, more large-scale clinical trials are needed. In the words of J McMurray,183 we might propose that ‘because ARBs are more costly than ACEis, their primary value is as an alternative for patients who cannot tolerate ACEis because of cough.’
Future of ACEis
Owing to the pivotal role of RAS in cardiovascular pathophysiology, there is continued interest in new compounds that can target this system. Of particular interest are the vasopeptidase inhibitors. These are dual ACE–neutral endopeptidase inhibitors, and omapatrilat is the most advanced in clinical development. Neutral endopeptidase, a zinc-metallopeptidase, degrades vasodilatory and diuretic natriuretic peptides that reduce volume loading and are therefore beneficial in both hypertension and heart failure.184 As expected, omapatrilat has shown better results than ACEis in clinical trials, but recent larger clinical trials have revealed a problematic incidence of angioedema with omapatrilat.185, 186 Both ACE and neutral endopeptidase inhibit bradykinin degradation, and bradykinin has been implicated in the angioedema associated with ACE inhibition.187 Therefore, dual ACE–neutral endopeptidase inhibition may result in a higher incidence of angioedema.188
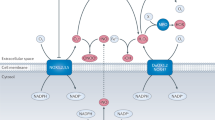
For these reasons, single-domain inhibitors of ACE offer an attractive alternative. The N- and C-domain sites of ACE hydrolyze Ang I and BK at comparable rates in vitro, but in vivo it seems that the C-domain is primarily responsible for regulating BP.189 Ang I is hydrolyzed predominantly by the C-domain in vivo,189 whereas BK is hydrolyzed by both domains.190 Therefore, selective inhibition of the C-domain site will allow some level of BK degradation to continue, catalyzed by the N-domain. This may prevent excessive BK accumulation during angioedema.191 Moreover, a C-selective inhibitor will have a lower propensity for excessive B2 receptor stimulation by BK, which is maximal when both the N- and C-domains are inhibited.192 Therefore, a highly selective C-domain inhibitor has the potential for effective BP control with reduced vasodilator-related side effects.
In contrast to a C-selective inhibitor, an N-selective inhibitor might open up new therapeutic areas. The N-domain has a minor role in BP control in vivo. At least three physiologically important peptides are hydrolyzed preferentially or exclusively by the N-domain: luteinizing hormone releasing hormone, Ang 1–7 and AcSDKP (acetyl-Ser-Asp-Lys-Pro).193, 194, 195 There is increasing evidence that ACE is the principal metabolizing enzyme for AcSDKP, a natural hemoregulatory hormone.196 AcSDKP has antiproliferative and antifibrotic activities, and it protects hematopoietic stem cells against chemotherapy-induced injury196 and limits cardiac fibrosis.197
The current-generation ACEis in clinical use are essentially mixed N- and C-domain inhibitors.198 Captopril has been noted to be modestly N-selective depending on Cl− concentration, whereas lisinopril and enalaprilat are more C-selective194, 199 (Figure 3). Keto-ACE, originally described in 1980, was found to exhibit a 40- to 50-fold C-selectivity.195 One of the bradykinin potentiator peptides, BPPb, was shown to be 300-fold more C-selective,200 and the phosphinic tetrapeptide RXPA380 is 3000-fold more C-selective.190 However, BPP-12b is 30-fold more N-selective, and the phosphinic tetrapeptide RXP407 is 1000-fold more N-selective201, 202, 203 (Figure 3).

Structures of different angiotensin-converting enzyme (ACE) inhibitors: (a) captopril, (b) lisinopril, (c) keto-ACE, (d) RXP407 and (e) RXPA380.
Conclusion
A few years ago, it was thought that we knew all about RAS and much about its regulation. However, the clinical applications of ACEis are widening from cardiovascular to other related morbidities. Improvement of the patient's cardiovascular risk by blockade of the RAS is caused primarily by BP reduction, but additional nonhemodynamic effects contribute as well. The beneficial effects of ACEis have gone beyond ACE inhibition, and their role in noncardiac functions is also strengthening. New aspects of the RAS, such as ACE 2, continue to emerge and could become targets for new therapeutic strategies. These new facets of RAAS will continue to fuel the interest of researchers to explore this system further.
Conflict of interest
The authors declare no conflict of interest.
References
Levy D, Larson MG, Vasan RS, Kannel WB, Ho KKL . The progression from hypertension to congestive heart failure. JAMA 1996; 275: 1557–1562.
http://www.mayoclinic.com/health/high-blood-pressure/HI00062.
Lonn E, Gerstein HC, Smieja M, Mann JFE, Yusuf S . Mechanisms of cardiovascular risk reductions with ramipril: insights from HOPE and HOPE substudies. Eur Heart J 2003; 5 (Suppl A): A43–A48.
Bicket DP . Using ACE inhibitors appropriately. Am Fam Physician 2002; 66: 461–468.
Skidgel RA, Erdos E . Biochemistry of angiotensin I-converting enzyme. In: Robertson JIS, Nicholls MG (eds). The Renin-Angiotensin System. Raven Press Ltd: New York, 1993. pp 1.01–10.10.
Brown NJ, Vaughan DE . Angiotensin-converting enzyme inhibitors. Circulation 1998; 97: 1411–1420.
Chobian AV, Barkis GL, Black HR, Cushman WC, Green LA, Izzo Jr JL, Jones DW, Materson BJ, Oparil S, Wright Jr JT, Roccella EJ . The seventh report of the joint national committee on prevention, detection, evaluation and treatment of high blood pressure. JAMA 2003; 289: 2560–2571.
ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Anti-hypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002; 288: 2981–2997.
Vidt DG, Bravo EL, Fouad FM . Medical intelligence therapy: captopril. N Engl J Med 1982; 306: 214–219.
Todd PA, Heel RC . Enalapril: a review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in hypertension and congestive heart failure. Drugs 1986; 31: 198–248.
Pool JL, Gennari J, Goldstein R, Kochar MS, Lewin AJ, Maxwell MH, McChesney JA, Mehta J, Nash DT, Nelson EB, Rastogi S, Rofman B, Weinberger M . Controlled multicenter study of antihypertensive effects of lisinopril, hydrochlorothiazide, and lisinopril plus hydrochlorothiazide in the treatment of 394 patients with mild to moderate essential hypertension. J Cardiovasc Pharmacol 1987; 3: S36–S42.
Case DB, Atlas SA, Laragh JH, Sullivan PA, Sealey JE . Use of first-dose response or plasma renin activity to predict the long-term effect of captopril: identification of triphasic pattern of blood pressure response. J Cardiovasc Pharmacol 1980; 2: 339–346.
Given BD, Taylor T, Hollenberg NK, Williams GH . Duration of action and short-term hormonal responses to enalapril (MK-421) in normal subjects. J Cardiovasc Pharmacol 1984; 6: 436–441.
Waeber B, Gavras I, Brunner HR, Cook CA, Charocopos F, Gavras HP . Prediction of sustained antihypertensive efficacy of chronic captopril therapy: relationships to immediate blood pressure response and control plasma renin activity. Am Heart J 1982; 103: 384–390.
Waeber B, Brunner HR, Brunner DB, Curtet AL, Turini CA, Gavras H . Discrepancy between antihypertensive effect and angiotensin converting enzyme inhibition by captopril. Hypertension 1980; 2: 236–242.
Veterans Administration Co-operative Study Group of Antihypertensive Agents. Racial differences in response to low-dose captopril are abolished by the addition of hydrochlorothiazide. Br J Clin Pharmacol 1982; 14: 97S–101S.
Lund-Johansen P, Omvik P . Long-term haemodynamic effects of enalapril (alone and in combination with hydrochlorothiazide) at rest and during exercise in essential hypertension. J Hypertens 1984; 2: S49–S50.
Ibsen H, Egan B, Osterzeil K, Vander A, Julius S . Reflex-hemodynamic adjustments and baroreflex sensitivity during converting enzyme inhibition with MK-421 in normal humans. Hypertension 1983; 5 (Suppl I): I–184–I-189.
Dunn GF, Oigman W, Ventura HO, Messerli FH, Kobrin I, Frolich ED . Enalapril improves systemic and renal hemodynamics and allows regression of left ventricular mass in essential hypertension. Am J Cardiol 1984; 53: 105–108.
Simon AC, Levenson JA, Bouthier J, Maarek B, Safar ME . Effects of acute and chronic angiotensin enzyme inhibition on large arteries in human hypertension. J Cardiovasc Pharmacol 1985; 7: S45–S51.
Dusing R, Kayser G, Wagner S, Scherf H, Glanzer K, Predel HG, Kramer HJ . Baroreflex setting and sensitivity in normal subjects: effects of pharmacologic inhibition of the angiotensin I converting enzyme. Am J Cardiol 1987; 59: 50D–54D.
Guidicelli JF, Berdeaux A, Edouard A, Richer C, Jacolot D . The effect of enalapril on baroreceptor mediated reflex function in normotensive subjects. Br J Clin Pharmacol 1985; 20: 211–218.
Ajayi AA, Campbell BC, Meredith PA, Kelman AW, Reid JL . The effect of captopril on the reflex control heart rate: possible mechanisms. Br J Clin Pharmacol 1985; 20: 17–25.
Giannattasio C, Grassi G, Seravalle G, Morganti A, Zanchetti A, Mancia G . Investigation of reflexes from volume and baroreceptors during converting-enzyme inhibition in humans. Am Heart J 1989; 117: 740–745.
Uchiyama-Tanaka Y, Mori Y, Kishimoto N, Fukui M, Nose A, Kijima Y, Yamahara H, Hasegawa T, Kosaki A, Matsubara H, Iwasaka T . Comparison of the effects of quinapril and losartan on carotid artery intima-media thickness in patients with mild-to-moderate arterial hypertension. Kidney Blood Press Res 2005; 28: 111–116.
Hayek T, Keidar S, Mei-Y, Oiknine J, Breslow J . Effect of angiotensin converting enzyme inhibitors on LDL lipid peroxidation and atherosclerosis progression in apo E deficient mice. Circulation 1995; 92 (Suppl I): I–625.
Chobanian AV, Haudenschild CC, Nickerson C, Drago R . Antiatherogenic effect of captopril in the Watanabe heritable hyperlipidemic rabbit. Hypertension 1990; 15: 327–331.
Aberg G, Ferrer P . Effects of captopril on atherosclerosis in cynomolgus monkeys. J Cardiovasc Pharmacol 1990; 15 (suppl 5): S65–S72.
Morawietz H, Rueckschloss U, Niemann B, Duerrschmidt N, Galle J, Hakim K, Zerkowski HR, Sawamura T, Holtz J . Angiotensin II induces LOX-1, the human endothelial receptor for oxidized low-density lipoprotein. Circulation 1999; 100: 899–902.
Mancini GB, Henry GC, Macaya C, O'Neill BJ, Pucillo AL, Carere RG, Wargovisch TJ, Mudra H, Luscher TF, Klibaner MI, Haber HE, Uprichard AC, Pepine CJ, Pitt B . Angiotensin-converting enzyme inhibition with quinapril improves endothelial vasomotor dysfunction in patients with coronary artery disease: the TREND (Trial on Reversing Endothelial Dysfunction) Study. Circulation 1996; 94: 258–265.
Linz W, Wiemer G, Gohlke P, Unger T, Scholkens BA . Contribution of kinins to the cardiovascular actions of angiotensin-converting enzyme inhibitors. Pharmacol Rev 1995; 47: 25–49.
Vaughan DE, Lazos SA, Tong K . Angiotensin II regulates the expression of plasminogen activator inhibitor-1 in cultured endothelial cells. J Clin Invest 1995; 95: 995–1001.
Ridker PM, Gaboury CL, Conlin PR, Seely EW, Williams GH, Vaughan DE . Stimulation of plasminogen activator inhibitor in vivo by infusion of angiotensin II. Circulation 1993; 87: 1969–1973.
Emeis JJ, Tranquille N . On the role of bradykinin in secretion from vascular endothelial cells. Agents Actions 1992; 38: 285–291.
Brown NJ, Nadeau J, Vaughan DE . Selective stimulation of tissue-type plasminogen activator (t-PA) in vivo by infusion of bradykinin. Thromb Haemost 1997; 77: 522–525.
The Heart Outcomes Prevention Evaluation Study Investigators. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med 2000; 342: 145–153.
de Nigris F, D'Armiento FP, Somma P, Casini A, Andreini I, Sarlo F, Mansueto G, De Rosa G, Bonaduce D, Condorelli M, Napoli C . Chronic treatment with sulfhydryl angiotensin-converting enzyme inhibitors reduce susceptibility of plasma LDL to in vitro oxidation, formation of oxidation-specific epitopes in the arterial wall, and atherogenesis in apolipoprotein E knockout mice. Int J Cardiol 2001; 81: 107–115.
CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure: results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N Engl J Med 1987; 316: 1429–1435.
The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med 1991; 325: 293–302.
Cohn JN, Johnson G, Ziesche J, Frederick C, Francis G, Tristani F, Smith R, Dunkman WB, Loeb H, Wong M, Bhat G, Goldman S, Fletcher RD, Doherty J, Hughes CV, Carson P, Cintron G, Shabetai R, Haakenson C . A comparison of enalapril with hydralazine-isosorbidedinitrate in the treatment of chronic congestive heart failure. N Engl J Med 1991; 325: 303–310.
AIRE Study Group. Effect of ramipril on mortality and morbidity of survivors of acute myocardial infarction with clinical evidence of heart failure. Lancet 1993; 342: 821–828.
Kober L, Torp-Pederson C, Carlsen JE, Bagger H, Eliasen P, Lyngborg K, Videbaek J, Cole DS, Auclert L, Pauly NC, Aliot E, Persson S, Camm AJ . For the Trandolapril Cardiac Evaluation (TRACE) Study Group. A clinical trial of the angiotensin-converting-enzyme inhibitor trandolapril in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 1995; 333: 1670–1676.
Pfeffer MA, Braunwald EA, Moye LA, Basta L, Brown EJJ, Cuddy TE, Davis BR, Geltman EM, Goldman S, Flaker GC, Klein M, Lamas GA, Packer M, Rouleau J, Rouleau JL, Rutherford J, Wertheimer JH, Hawkins CM . Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 1992; 327: 669–677.
Yusuf S, Pepine CJ, Garces C, Pouleur H, Salem D, Kostis J, Benedict C, Rousseau M, Bourassa M, Pitt B . Effect of enalapril on myocardial infarction and unstable angina in patients with low ejection fractions. Lancet 1992; 340: 1173–1178.
Vaughan DE, Pfeffer MA . Angiotensin converting enzyme inhibitors and cardiovascular remodelling. Cardiovasc Res 1994; 28: 159–165.
Dahlof B . Effect of angiotensin II a blockade on cardiac hypertrophy and remodelling: a review. J Hum Hypertens 1995; 9 (suppl 5): S37–S44.
Nakashima Y, Fouad FM, Tarazi RC . Regression of left ventricular hypertrophy from systemic hypertension by enalapril. Am J Cardiol 1994; 53: 1044–1049.
Gaudio C, Tanzilli G, Collatina S, Pagnotta P, Paknejad K, Campa PP . Evaluation of regression of left ventricular hypertrophy in hypertensive patients treated with captopril as assessed by magnetic resonance imaging. Cardiologia 1992; 37: 789–791.
Schmieder RE, Martus P, Klingbeil A . Reversal of left ventricular hypertrophy in essential hypertension: meta-analysis of randomized double-blind studies. JAMA 1996; 275: 1507–1513.
Givertz MM . Manipulation of the renin–angiotensin system. Circulation 2001; 104: e14–e18.
ACE Inhibitor Myocardial Infarction Collaborative Group. Indications for ACE inhibitors in the early treatment of acute myocardial infarction: systematic overview of individual data from 100 000 patients in randomized trials. Circulation 1998; 97: 2202–2212.
J S . Angiotensin-converting enzyme inhibition: a landmark advance in treatment for cardiovascular diseases. Eur Heart J Suppl 2007; 9 (Supplement E): E2–E9.
Ferrari R . Effects of angiotensin-converting enzyme inhibition with perindopril on left ventricular remodeling and clinical outcome: results of the randomized Perindopril and Remodeling in Elderly with Acute Myocardial Infarction (PREAMI) Study. Arch Intern Med 2006; 166: 659–666.
Ryan TJ, Antman EM, Brooks NH, Califf RM, Hillis LD, Hiratzka LF, Rapaport E, Riegel B, Russell RO, Smith 3rd EE, Weaver WD, Gibbons RJ, Alpert JS, Eagle KA, Gardner TJ, Garson Jr A, Gregoratos G, Smith Jr SC . 1999 update: ACC/AHA guidelines for the management of patients with acute myocardial infarction: executive summary and recommendations: a report of the American college of cardiology/American heart association task force on practice guidelines (committee on management of acute myocardial infarction). Circulation 1999; 100: 1016–1030.
Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G . Effects of an angiotensin-converting enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients: the Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med 2000; 342: 145–153.
Ichikawa I, Brenner BM . Glomerular actions of angiotensin II. Am J Med 1984; 76: 43–49.
Fogo A, Yoshida Y, Yared A, Ichikawa I . Importance of angiogenic action of angiotensin II in the glomerular growth of maturing kidneys. Kidney Int 1990; 38: 1068–1074.
Ruiz-Ortega M, Gomez-Garre D, Alcazar R, Palacios I, Bustos C, Gonzalez S, Plaza JJ, Gonzalez E, Egido J . Involvement of angiotensin II and endothelin in matrix protein production and renal sclerosis. J Hypertens 1994; 12: S51–S58.
Anderson SG, Rennke HG, Brenner BM . Therapeutic advantage of converting enzyme inhibitors in arresting progressive renal disease associated with systemic hypertension in rat. J Clin Invest 1986; 77: 1993–2000.
Hoelscher DD, Weir MR, Bakris GL . Hypertension in diabetic patients: an update of interventional studies to preserve renal function. J Clin Pharmacol 1995; 35: 73–80.
Ravid M, Lang R, Rachmani R, Lishner M . Long-term renoprotective effect of angiotensin-converting enzyme inhibition in non-insulin dependent diabetes mellitus: a 7-year follow-up study. Arch Intern Med 1996; 156: 286–289.
Lewis EJ, Hunsicker LG, Bain RP, Rohde RD . The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. N Engl J Med 1993; 329: 1456–1462.
Maschio G, Alberti D, Janin G, Locatelli F, Mann JF, Motolese M, Ponticelli C, Ritz E, Zucchelli P . Effect of the angiotensin-converting enzyme inhibitor benazepril on the progression of chronic renal insufficiency: the Angiotensin-Converting-Enzyme Inhibition in Progressive Renal Insufficiency Study Group. N Engl J Med 1996; 334: 939–945.
Amann B, Tinzmann R, Angelkort B . ACE Inhibitors improve diabetic nephropathy through suppression of renal MCP-1. Diabetes Care 2003; 26: 2421–2425.
Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK . Angiotensin II stimulation of NAD(P)H oxidase activity. Circ Res 2002; 91: 406–413.
Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M, Li H, Bodenschatz M, August M, Kleschyov AL, Tsilimingas N, Walter U, Forstermann U, Meinertz T, Griendling K, Munzel T . Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res 2002; 90: E58–E65.
Wang HD, Xu S, Johns DG, Du Y, Quinn MT, Cayatte AJ, Cohen R . Role of NADPH oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice. Circ Res 2001; 88: 947–953.
Escobales N, Crespo MJ . Oxidative-nitrosative stress in hypertension (Review). Curr Vasc Pharmacol 2005; 3: 231–246.
Cotter MA, Cameron NE . Effect of the NAD(P)H oxidase inhibitor, apocynin, on peripheral nerve perfusion and function in diabetic rats. Life Sci 2003; 73: 1813–1824.
Aggarwal M, Singh J, Sood S, Arora B . Effects of lisinopril on streptozotocin-induced diabetic neuropathy in rats. Methods Find Exp Clin Pharamacol 2001; 23: 131–134.
Malik RA, Williamson S, Abbott C, Carrington AL, Iqbal J, Schady W, Boulton AJ . Effect of angiotensin-converting-enzyme (ACE) inhibitor trandolapril on human diabetic neuropathy: randomized double-blind controlled trial. Lancet 1998; 352: 1978–1981.
Reja A, Tesfaye S, Harris ND, Ward JD . Is ACE inhibition with lisinopril helpful in diabetic neuropathy? Diabet Med 1995; 12: 307–309.
Terata K, Coppey LJ, Davidson EP, Dunlap JA, Gutterman DD, Yorek MA . Acetylcholine-induced arteriolar dilation is reduced in streptozotocin (STZ)-induced diabetic rats with motor nerve dysfunction. Br J Pharmacol 1999; 128: 837–843.
Yorek MA, Coppey LJ, Gellett JS, Davidson EP . Sensory nerve innervations of epineurial arterioles of the sciatic nerve containing calcitonin gene related peptide: effect of streptozotocin-induced diabetes. Exp Diabesity Res 2004; 5: 187–193.
Coppey LJ, Gellett JS, Davidson EP, Yorek MA . Preventing superoxide formation in epineurial arterioles of the sciatic nerve from diabetic rats restores endothelium-dependent vasodilation. Free Radical Res 2003; 37: 33–40.
Coppey LJ, Gellett JS, Yorek MA . Mediation of vascular relaxation in epineurial arterioles of the sciatic nerve: effect of diabetes in type 1 and type 2 diabetic rat models. Endothelium 2003; 10: 89–94.
Coppey L, Gellett J, Davidson E, Dunlap J, Lund D, Yorek M . Effect of antioxidant treatment of streptozotocin-induced diabetic rats on endoneurial blood flow, motor nerve conduction velocity, and vascular reactivity of epineurial arterioles of the sciatic nerve. Diabetes 2001; 50: 1927–1937.
Coppey LJ, Davidson EP, Rinehart TW, Gellett JS, Oltman CL, Lund DD, Yorek MA . ACE inhibitor or angiotensin II receptor antagonist attenuates diabetic neuropathy in streptozotocin-induced diabetic rats. Diabetes 2006; 55: 341–348.
O'Driscoll G, Green D, Rankin J, Stanton K, Taylor R . Improvement in endothelial function by angiotensin converting enzyme inhibition in insulin-dependent diabetes mellitus. J Clin Invest 1997; 100: 678–684.
Malik RA, Schofield IJ, Izzard A, Austin C, Bermann G, Heagerty AM . Effects of angiotensin type-1 receptor antagonism on small artery function in patients with type 2 diabetes mellitus. Hypertension 2005; 45: 264–269.
Goto K, Fujii K, Kansui Y, Iida M . Changes in endothelium-derived hyperpolarizing factor in hypertension and ageing: response to chronic treatment with rennin-angiotensin system inhibitors. Clin Exp Pharmacol Physiol 2004; 31: 650–655.
Kihara M, Mitsui MK, Mitsui Y, Okuda K, Nakasaka Y, Takahashi M, Schmelzer JD . Altered vasoreactivity to angiotensin II in experimental diabetic neuropathy: role of nitric oxide. Muscle Nerve 1999; 22: 920–925.
Griendling KK, Sorescu D, Ushio-Fukai M . NAD (P) H oxidase: role in cardiovascular biology and disease. Circ Res 2000; 86: 494–501.
Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW . Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 1994; 74: 1141–1148.
Laursen JB, Rajagopalan S, Galis Z, Tarpey M, Freeman BA, Harrison DG . Role of superoxide in angiotensin II–induced but not catecholamine-induced hypertension. Circulation 1997; 95: 588–593.
Zafari AM, Ushio-Fukai M, Akers M, Yin Q, Shah A, Harrison DG, Taylor WR, Griendling KK . Role of NADH/NADPH oxidase–derived H2O2 in angiotensin II–induced vascular hypertrophy. Hypertension 1998; 32: 488–495.
Warnholtz A, Nickenig G, Schulz E, Macharzina R, Bräsen JH, Skatchkov M, Heitzer T, Stasch JP, Griendling KK, Harrison DG, Böhm M, Meinertz T, Münzel T . Increased NADH oxidase–mediated superoxide production in the early stages of atherosclerosis: evidence for involvement of the renin-angiotensin system. Circulation 1999; 99: 2027–2033.
Münzel T, Keaney JF . Are ACE inhibitors a ‘Magic Bullet’ against oxidative stress? Circulation 2001; 104: 1571.
Mancini GB, Henry GC, Macaya C, O'Neill BJ, Pucillo AL, Carere RG, Wargovich TJ, Mudra H, Lüscher TF, Klibaner MI, Haber HE, Uprichard AC, Pepine CJ, Pitt B . Angiotensin-converting enzyme inhibition with quinapril improves endothelial vasomotor dysfunction in patients with coronary artery disease: the TREND (Trial on Reversing Endothelial Dysfunction) Study. Circulation 1996; 94: 258–265.
Wiemer G, Linz W, Hatrik S, Schölkens BA, Malinski T . Angiotensin-converting enzyme inhibition alters nitric oxide and superoxide release in normotensive and hypertensive rats. Hypertension 1997; 30: 1183–1190.
Lonn E, Yusuf S, Dzavik V, Doris C, Yi Q, Smith S, Moore-Cox A, Bosch J, Riley W, Teo K, SECURE Investigators. Effects of ramipril and vitamin E on atherosclerosis: the study to evaluate carotid ultrasound changes in patients treated with ramipril and vitamin E (SECURE). Circulation 2001; 103: 919–925.
Napoli C, Cicala C, D'Armiento FP, Roviezzo F, Somma P, de Nigris F, Zuliani P, Bucci M, Aleotti L, Casini A, Franconi F, Cirino G . Beneficial effects of ACE-inhibition with zofenopril on plaque formation and low-density lipoprotein oxidation in watanabe heritable hyperlipidemic rabbits. Gen Pharmacol 1999; 33: 467–477.
Fernandes AC, Filipe PM, Freitas JP, Manso CF . Different effects of thiol and nonthiol ACE inhibitors on copper-induced lipid and protein oxidative modification. Free Radic Biol Med 1996; 20: 507–514.
Kim JA, Gu JL, Natarajan R, Berliner JA, Nadler JL . A leukocyte type of 12-lipoxygenase is expressed in human vascular and mononuclear cells. Evidence for upregulation by angiotensin II. Arterioscler Thromb Vasc Biol 1995; 15: 942–948.
Scheidegger KJ, Butler S, Witztum JL . Angiotensin II increases macrophage-mediated modification of low density lipoprotein via a lipoxygenase-dependent pathway. J Biol Chem 1997; 272: 21609–21615.
Keidar S, Kaplan M, Hoffman A, Aviram M . Angiotensin II stimulates macrophage-mediated oxidation of low density lipoproteins. Atherosclerosis 1995; 115: 201–215.
Parthasarathy S, Wieland E, Steinberg D . A role for endothelial cell lipoxygenase in the oxidative modification of low density lipoprotein. Proc Natl Acad Sci USA 1989; 86: 1046–1050.
Dohi Y, Hahn AWA, Boulanger CM, Buhler FR, Luscher TF . Endothelin stimulated by angiotensin II augments contractility of spontaneously hypertensive rat resistance arteries. Hypertension 1992; 19: 131–137.
Luscher TF, Boulanger CM, Dohi Y, Yang Z . Endothelium derived contracting factors. Hypertension 1992; 19: 117–130.
Turlapaty PDMV, Altura BM . Magnesium deficiency produces spasms of the coronary arteries: relationships to etiology of sudden ischemic heart disease. Science 1980; 208: 198–200.
Eisenberg MJ . Magnesium deficiency and sudden death. Am Heart J 1992; 124: 544–549.
Sapna S, Ranjith SK, Shivakumar K . Cardiac fibrogenesis in magnesium deficiency: a role for circulating angiotensin II and aldosterone. Am J Physiol Heart Circ Physiol 2006; 291: H436–H440.
Mogielnicki A, Chabielska E, Pawlak R, Szemraj J, Buczko W . Angiotensin II enhances thrombosis development in renovascular hypertensive rats. Thromb Haemost 2005; 93: 1069–1076.
Ambrose JA, Martinez EE . A new paradigm for plaque stabilization. Circulation 2002; 105: 2000–2004.
Barbagallo M, Dominguez LJ, Resnick LM . Protective effects of captopril against ischemic stress. Hypertension 1999; 34: 958–963.
Amenta F, Mignini F, Rabbia F, Tomassoni D, Veglio F . Protective effect of anti-hypertensive treatment on cognitive function in essential hypertension: analysis of published clinical data. J Neurol Sci 2002; 203–204: 147–151.
Manschot SM, Biessels GJ, Cameron NE, Cotter MA, Kamal A, Kappelle LJ, Gispen WH . Angiotensin converting enzyme inhibition partially prevents deficits in water maze performance, hippocampal synaptic plasticity and cerebral blood flow in streptozotocin-diabetic rats. Brain Res 2003; 966: 274–282.
Wyss JM, Kadish I, van Groen T . Age-related decline in spatial learning and memory: attenuation by captopril. Clin Exp Hypertens 2003; 25: 455–474.
PROGRESS Collaborative Group. Randomised trial of a perindopril-based blood-pressure-lowering regimen among 6105 individuals with previous stroke or transient ischaemic attack. Lancet 2001; 358: 1033–1041.
Skidgel RA, Defendini R, Erdos EG . Angiotensin I converting enzyme and its role in neuropeptide metabolism. In Turner AJ (ed). Neuropeptides and their peptidases. Ellis Horwood Ltd: Chichester, UK, 1987. pp 165–182.
Hasenohrl RU, Souza-Silv AMA, Nikolaus S, Tomaz C, Brandao ML, Schwarting RK, Huston JP . Substance P and its role in neural mechanisms governing learning, anxiety and functional recovery. Neuropeptides 2000; 34: 272–280.
Oitzl MS, Hasenohrl RU, Huston JP . Reinforcing effects of peripherally administered substance P and its C-terminal sequence pGlu6-SP6-11 in the rat. Psychopharmacology (Berl) 1990; 100: 308–315.
Shirayama Y, Mitsushio H, Takashima M, Ichikawa H, Takahashi K . Reduction of substance P after chronic antidepressants treatment in the striatum, substantia nigra and amygdala of the rat. Brain Res 1996; 739: 70–78.
Tomaz C, Silva AC, Nogueira PJ . Long-lasting mnemotropic effect of substance P and its N-terminal fragment (SP1-7) on avoidance learning. Braz J Med Biol Res 1997; 30: 231–233.
Ukai M, Kobayashi T, Kameyama T . Dynorphin A-(1–13) attenuates basal forebrain-lesion-induced amnesia in rats. Brain Res 1993; 625: 355–356.
Hiramatsu M, Watanabe E . Dynorphin A (2–13) improves mecamylamine-induced learning impairment accompanied by reversal of reductions in acetylcholine release in rats. Neuropeptides 2006; 40: 47–56.
Hiramatsu M, Mori H, Murasawa H, Kameyama T . Improvement by dynorphin A (1–13) of galanin-induced impairment of memory accompanied by blockade of reductions in acetylcholine release in rats. Br J Pharmacol 1996; 118: 255–260.
Gold PE . Coordination of multiple memory systems. Neurobiol Learn Mem 2004; 82: 230–242.
Gold PE . Acetylcholine modulation of neural systems involved in learning and memory. Neurobiol Learn Mem 2003; 80: 194–210.
Ager EI, Neo J, Christophi C . The renin–angiotensin system and malignancy. Carcinogenesis 2008; 29: 1675–1684.
Deshayes F, Nahmias C . Angiotensin receptors: a new role in cancer? Trends Endocrinol Metab 2005; 16: 293–299.
Escobar E, Rodríguez-Reyna TS, Arrieta O, Sotelo J . Angiotensin II, cell proliferation and angiogenesis regulator: biologic and therapeutic implications in cancer. Curr Vasc Pharmacol 2004; 2: 385–399.
Uemura H, Ishiguro H, Kubota Y . Angiotensin II receptor blocker: possibility of antitumor agent for prostate cancer. Mini Rev Med Chem 2006; 6: 835–844.
Zhao Y, Chen X, Cai L, Yang Y, Sui G, Wu J . Angiotensin II suppresses adriamycin-induced apoptosis through activation of phosphatidylinositol 3-kinase/Akt signaling in human breast cancer cells. Acta Biochim Biophys Sin (Shanghai) 2008; 40: 304–310.
Ohashi H, Takagi H, Oh H, Suzuma K, Suzuma I, Miyamoto N, Uemura A, Watanabe D, Murakami T, Sugaya T, Fukamizu A, Honda Y . Phosphatidylinositol 3-kinase/Akt regulates angiotensin II-induced inhibition of apoptosis in microvascular endothelial cells by governing survivin expression and suppression of caspase-3 activity. Circ Res 2004; 94: 785–793.
Puddefoot JR, Udeozo UKI, Barker S, Vinson GP . The role of angiotensin II in the regulation of breast cancer cell adhesion and invasion. Endocr Relat Cancer 2006; 13: 895–903.
Ribatti D, Conconi MT, Nussdorfer GG . Nonclassic endogenous novel regulators of angiogenesis. Pharmacol Rev 2007; 59: 185–205.
Fujita M, Hayashi I, Yamashina S, Fukamizu A, Itoman M, Majima M . Angiotensin type 1a receptor signaling-dependent induction of vascular endothelial growth factor in stroma is relevant to tumor-associated angiogenesis and tumor growth. Carcinogenesis 2005; 26: 271–279.
Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J . Inflammation and angiotensin II. Int J Biochem Cell Biol 2003; 35: 881–900.
El Bekay R, Alvarez M, Monteseirín J, Alba G, Chacón P, Vega A, Martin-Nieto J, Jiménez J, Pintado E, Bedoya FJ, Sobrino F . Oxidative stress is a critical mediator of the angiotensin II signal in human neutrophils: involvement of mitogen activated protein kinase, calcineurin, and the transcription factor NF-kB. Blood 2003; 102: 662–671.
Hii SI, Nicol DL, Gotley DC, Thompson LC, Green MK, Jonsson JR . Captopril inhibits tumour growth in a xenograft model of human renal cell carcinoma. Br J Cancer 1998; 77: 880–883.
Volpert OV, Ward WF, Lingen MW, Chesler L, Solt DB, Johnson MD, Molteni A, Polverini PJ, Bouck NP . Captopril inhibits angiogenesis and slows the growth of experimental tumors in rats. J Clin Invest 1996; 98: 671–679.
Yoshiji H, Kuriyama S, Kawata M, Yoshii J, Ikenaka Y, Noguchi R, Nakatani T, Tsujinoue H, Fukui H . ACE inhibitor perindopril significantly inhibits tumor growth and angiogenesis along with suppression of the VEGF level. Clin Cancer Res 2001; 7: 1073–1078.
Lindberg H, Nielsen D, Jensen BV, Eriksen J, Skovsgaard T . Angiotensin converting enzyme inhibitors for cancer treatment? Acta Oncol 2004; 43: 142–152.
Williams RN, Parsons S, Rowlands B, Watson S . Novel inhibition of matrix metalloproteinases, angiogenesis, and tumour cell invasion by captopril. Gastroenterology 2003; 124: A603.
Williams RN, Parsons SL, Morris TM, Rowlands BJ, Watson SA . Inhibition of matrix metalloproteinase activity and growth of gastric adenocarcinoma cells by an angiotensin converting enzyme inhibitor in in vitro and murine models. Eur J Surg Oncol 2005; 31: 1042–1050.
de Groot-Besseling RR, Ruers TJ, van Kraats AA, Poelen GJ, Ruiter DJ, de Waal RM, Westphal JR . Anti-tumor activity of a combination of plasminogen activator and captopril in a human melanoma xenograft model. Int J Cancer 2004; 112: 329–334.
McMurray J, Chopra M . Influence of ACE inhibitors on free radicals and reperfusion injury: pharmacological curiosity or therapeutic hope? Br J Clin Pharmacol 1991; 31: 373–379.
Wysocki PJ, Kwiatkowska EP, Kazimierczak U, Suchorska W, Kowalczyk DW, Mackiewicz A . Captopril, an angiotensin-converting enzyme inhibitor, promotes growth of immunogenic tumors in mice. Clin Cancer Res 2006; 12: 4095–4102.
Friis S, Sørensen HT, Mellemkjaer L, McLaughlin JK, Nielsen GL, Blot WJ, Olsen JH . Angiotensin-converting enzyme inhibitors and the risk of cancer: a population-based cohort study in Denmark. Cancer 2001; 92: 2462–7240.
Meier CR . Angiotensin-converting enzyme inhibitors, calcium channel blockers, and breast cancer. Arch Intern Med 2000; 160: 349–355.
Grossman E, Messerli FH, Goldbourt U . Antihypertensive therapy and the risk of malignancies. Eur Heart J 2001; 22: 1343–1352.
Lever AF, Hole DJ, Gillis CR, McCallum IR, McInnes GT, MacKinnon PL, Meredith PA, Murray LS, Reid JL, Robertson JW . Do inhibitors of angiotensin-converting enzyme protect against risk of cancer? Lancet 1998; 352: 179–184.
Silber JH, Cnaan A, Clark BJ, Paridon SM, Chin AJ, Rychik J, Hogarty AN, Cohen MI, Barber G, Rutkowski M, Kimball TR, Delaat C, Steinherz LJ, Zhao H . Enalapril to prevent cardiac function decline in long-term survivors of pediatric cancer exposed to anthracyclines. J Clin Oncol 2004; 22: 820–822.
Medeiros R, Vasconcelos A, Costa S, Pinto D, Lobo F, Morais A, Oliveira J, Lopes C . Linkage of angiotensin I-converting enzyme gene insertion/deletion polymorphism to the progression of human prostate cancer. J Pathol 2004; 202: 330–335.
Rocken C, Lendecke U, Dierkes J, Westphal S, Carl-McGrath S, Peters B, Kruger S, Malfertheiner P, Roessner A, Ebert MPA . The number of lymph node metastases in gastric cancer correlates with the angiotensin I converting enzyme gene insertion/deletion polymorphism. Clin Cancer Res 2005; 11: 2526–2530.
Ebert MP, Lendeckel U, Westphal S, Dierkes J, Glas J, Folwaczny C, Roessner A, Stolte M, Malfertheiner P, Röcken C . The Angiotensin I–converting enzyme gene insertion/deletion polymorphism is linked to early gastric cancer. Cancer Epidemiol Biomarkers Prev 2005; 14: 2987–2989.
Grand AM, Maresca AM . Blockade of the renin-angiotensin-aldosterone system: effects on hypertensive target organ damage. Cardiovasc Hematolo Agents Med Chem 2006; 4: 219–228.
Brilla CG, Janicki JS, Weber KT . Impaired diastolic function and coronary reserve in genetic hypertension. Role of interstitial fibrosis and medial thickening of intramyocardial coronary arteries. Circ Res 1991; 69: 107.
Anderson S, Rennke HG, Brenner BM . Therapeutic advantage of converting enzyme inhibitors in arresting progressive renal disease associated with systemic hypertension in the rat. J Clin Invest 1986; 77: 1993–2000.
Maschio G, Alberti D, Janin G, Locatelli F, Mann JF, Motolese M, Ponticelli C, Ritz E, Zucchelli P . Effect of the angiotensin-converting-enzyme inhibitor benazepril on the progression of chronic renal insufficiency. N Engl J Med 1996; 334: 939–945.
Schachinger V, Britten MB, Zeiher AM . Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation 2000; 101: 1899–1906.
Ghiadoni L, Magagna A, Versari D, Kardasz I, Huang Y, Taddei S, Salvetti A . Different effect of antihypertensive drugs on conduit artery endothelial function. Hypertension 2003; 41: 1281–1286.
Hecker M, Blaukat A, Bara AT, Müller-Esterl W, Busse R . ACE inhibitor potentiation of bradykinin-induced venoconstriction. Br J Pharmacol 1997; 121: 1475–1481.
Hecker M, Pörsti I, Bara AT, Busse R . Potentiation by ACE inhibitors of the dilator response to bradykinin in the coronary microcirculation: interaction at the receptor level. Br J Pharmacol 1994; 111: 238–244.
Minshall RD, Erdös EG, Vogel SM . Angiotensin I-converting enzyme inhibitors potentiate bradykinin's inotropic effects independently of blocking its inactivation. Am J Cardiol 1997; 80: 132A–136A.
Minshall RD, Tan F, Nakamura F, Rabito SF, Becker RP, Marcic B, Erdös EG . Potentiation of the actions of bradykinin by angiotensin I-converting enzyme inhibitors. The role of expressed human bradykinin B2 receptors and angiotensin I-converting enzyme in CHO cells. Circ Res 1997; 81: 848–856.
Ignjatovic T, Tan F, Brovkovych V, Skidgel RA, Erdös EG . Activation of bradykinin B1 receptor by ACE inhibitors. Int Immunopharmacol 2002; 2: 1787–1793.
Kohlstedt K, Busse R, Fleming I . Signaling via the angiotensin-converting enzyme enhances the expression of cyclooxygenase-2 in endothelial cells. Hypertension 2005; 45: 126–132.
Kohlstedt K, Shoghi F, Müller-Esterl W, Busse R, Fleming I . CK2 phosphorylates the angiotensin-converting enzyme and regulates its retention in the endothelial cell plasma membrane. Circ Res 2002; 91: 749–756.
Irvine RJ, White JM, Head RJ . The renin angiotensin system and nociception in spontaneously hypertensive rats. Life Sci 1995; 56: 1073–1078.
Ghione S . Hypertension-associated hypalgesia: evidence in experimental animals and humans, pathophysiological mechanisms, and potential clinical consequences. Hypertension 1996; 28: 494–504.
Takai S, Song K, Tanaka T, Okunishi H, Miyazaki M . Antinociceptive effects of angiotensin-converting enzyme inhibitors and an angiotensin II receptor antagonist in mice. Life Sci 1996; 59: PL331–PL336.
Guasti L, Grimoldi P, Diolisi A, Petrozzino MR, Gaudio G, Grandi AM, Rossi MG, Venco A . Treatment with enalapril modifies the pain perception pattern in hypertensive patients. Hypertension 1998; 31: 1146–1150.
Kalra J, Chaturvedi A, Kalra S, Chaturvedi H, Dhasmana DC . Modulation of pain perception by ramipril and losartan in human volunteers. Indian J Physiol Pharmacol 2008; 52: 91–96.
Brunton L, Parker K, Blumenthal D, Buxton I (eds). Renin and angiotensin. Goodman & Gillman's Manual of Pharmacology and Therapeutics. McGraw Hill: New York, 2008. pp 511–527.
Warren SE, O'Connor DT . Hyperkalemia resulting from captopril administration. JAMA 1980; 244: 2551–2552.
Textor SC, Bravo EL, Fouad FM, Tarazi RC . Hyperkalemia in azotemic patients during angiotensin-converting enzyme inhibition and aldosterone reduction with captopril. Am J Med 1982; 73: 719–725.
Burnakis TG, Mioduch HJ . Combined therapy with captopril and potassium supplementation: a potential for hyperkalemia. Arch Int Med 1984; 144: 2371–2372.
Dzau VJ . Renal effects of angiotensin-converting enzyme inhibition in cardiac failure. Am J Kidney Dis 1987; 10: 74–80.
Murphy BF, Whitworth JA, Kincaid-Smith P . Renal insufficiency with combinations of angiotensin converting enzyme inhibitors and diuretics. Br Med J Clin Res 1984; 288: 844–845.
Kastner PR, Hall JE, Guyton AC . Control of glomerular filtration rate: role of intrarenally formed angiotensin II. Am J Physiol 1984; 246: F897–F906.
Israili ZH, Hall WD . Cough and angioneurotic edema associated with angiotensin-converting enzyme inhibitor therapy. Ann Intern Med 1992; 117: 234–242.
Friedman JM . ACE inhibitors and congenital anomalies. N Engl J Med 2006; 354: 2498–2500.
DiBianco R . Adverse reactions with angiotensin converting enzyme inhibitors. Med Toxicol 1986; 1: 122–141.
Marin-Castaño ME, Schanstra JP, Neau E, Praddaude F, Pecher C, Ader JL, Girolami JP, Bascands JL . Induction of functional bradykinin b (1)-receptors in normotensive rats and mice under chronic Angiotensin-converting enzyme inhibitor treatment. Circulation 2002; 105: 627–632.
Blood Pressure Lowering Treatment Trialists' Collaboration, Turnbull F, Neal B, Pfeffer M, Kostis J, Algert C, Woodward M, Chalmers J, Zanchetti A, MacMahon S . Blood pressure-dependent and independent effects of agents that inhibit the renin-angiotensin system. J Hypertens 2007; 25: 951–958.
Strauss MH, Hall AS . Angiotensin receptor blockers may increase risk of myocardial infarction: unraveling the ARB-MI paradox. Circulation 2006; 114: 838–854.
Groth W, Blume A, Gohlke P, Unger T, Culman J . Chronic pretreatment with candesartan improves recovery from focal cerebral ischaemia in rats. J Hypertens 2003; 21: 2175–2182.
Li J, Culman J, Hortnagl H, Zhao Y, Gerova N, Timm M, Blume A, Zimmermann M, Seidel K, Dirnagl U, Unger T . Angiotensin AT2 receptor protects against cerebral ischemia-induced neuronal injury. FASEB J 2005; 19: 617–619.
Sigmund CD, Davisson RL . Targeting brain AT1 receptors by RNA interference. Hypertension 2006; 47: 145–146.
Fournier A, Messerli FH, Achard JM, Fernandez L . Cerebroprotection mediated by angiotensin II: a hypothesis supported by recent randomized clinical trials. J Am Coll Cardiol 2004; 43: 1343–1347.
McMurray JJ . ACE inhibitors in cardiovascular disease-unbeatable? N Engl J Med 2008; 358: 1615–1616.
Weber MA . Vasopeptidase inhibitors. Lancet 2001; 358: 1525–1532.
Coats AJS . Omapatrilat—the story of OVERTURE and OCTAVE. Int J Cardiol 2002; 86: 1–4.
Zanchi A, Maillard M, Burnier M . Recent clinical trials with omapatrilat: new developments. Curr Hypertens Rep 2003; 5: 346–352.
Cugno M, Nussberger J, Cicardi M, Agostoni A . Bradykinin and the pathophysiology of angioedema. Int Immunopharmacol 2003; 3: 311–317.
Campbell DJ . Vasopeptidase inhibition. A double-edged sword? Hypertension 2003; 41: 383–389.
Junot C, Gonzales MF, Ezan E, Cotton J, Vazeux G, Michaud A, Azizi M, Vassiliou S, Yiotakis A, Corvol P, Dive V . RXP 407, a selective inhibitor of the N-domain of angiotensin I-converting enzyme, blocks in vivo the degradation of hemoregulatory peptide acetyl-Ser-Asp-Lys-Pro with no effect on angiotensin I hydrolysis. J Pharmacol Exp Ther 2001; 297: 606–611.
Geordiadis D, Beau F, Czarny B, Cotton J, Yiotakis A, Dive V . Roles of the two active sites of somatic angiotensin-converting enzyme in the cleavage of angiotensin I and bradykinin. Circ Res 2003; 93: 148–154.
Messerli FH, Nussberger J . Vasopeptidase inhibition and angioedema. Lancet 2000; 356: 608–609.
Tom B, de Vries R, Saxena PR, Danser AHJ . Bradykinin potentiation by angiotensin-(1–7) and ACE inhibitors correlates with ACE C- and N-domain blockade. Hypertension 2001; 38: 95–99.
Rousseau A, Michaud A, Chauvet MT, Lenfant M, Corvol P . The hemoregulatory peptide N-acetyl-Ser-Asp-Lys- Pro is a natural and specific substrate of the N-terminal active site of human angiotensin-converting enzyme. J Biol Chem 1995; 270: 3656–3661.
Ehlers MRW, Riordan JF . Angiotensin-converting enzyme: zinc- and inhibitor-binding stoichiometries of the somatic and testis isozymes. Biochemistry 1991; 30: 7118–7126.
Deddish PA, Marcic B, Jackman HL, Wang HZ, Skidgel RA, Erdös EG . N-domain- specific substrate and C-domain inhibitors of angiotensin-converting enzyme: Angiotensin-(1–7) and keto-ACE. Hypertension 1998; 31: 912–917.
Azizi M, Junot C, Ezan E, Menard J . Angiotensin I-converting enzyme and metabolism of the haematological peptide N-acetyl-seryl-aspartyl-lysyl-proline. Clin Exp Pharmacol Physiol 2001; 28: 1066–1069.
Rhaleb NE, Peng H, Harding P, Tayeh M, LaPointe MC, Carretero OA . Effect of N-acetyl-seryl-aspartyl-lysylproline on DNA and collagen synthesis in rat cardiac fibroblasts. Hypertension 2001; 37: 827–832.
Menard J, Patchett AA . Angiotensin-converting enzyme inhibitors. Adv Prot Chem 2001; 56: 13–75.
Wei L, Clauser E, Alhenc-Gelas F, Corvol P . The two homologous domains of human angiotensin I-converting enzyme interact differently with competitive inhibitors. J Biol Chem 1992; 267: 13398–13405.
Cotton J, Hayashi MA, Cuniasse P, Vazeux G, Ianzer D, De Camargo AC, Dive V . Selective inhibition of the C-domain of angiotensin I converting enzyme by bradykinin potentiating peptides. Biochemistry 2002; 41: 6065–6071.
Hayashi MA, Murbach AF, Ianzer D, Portaro FC, Prezoto BC, Fernandes BL, Silveira PF, Silva CA, Pires RS, Britto LR, Dive V, Camargo AC . The C-type natriuretic peptide precursor of snake brain contains highly specific inhibitors of the angiotensin-converting enzyme. J Neurochem 2003; 85: 969–977.
Dive V, Cotton J, Yiotakis A, Michaud A, Vassiliou S, Jiracek J, Vazeux G, Chauvet MT, Cuniasse P, Corvol P . RXP 407, a phosphinic peptide, is a potent inhibitor of angiotensin I converting enzyme able to differentiate between its two active sites. Proc Natl Acad Sci USA 1999; 96: 4330–4335.
Acharya KR, Sturrock ED, Riordan JF, Ehlers MR . ACE revisited: a new target for structure-based drug design. Nat Rev Drug Discov 2003; 2: 891–902.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hanif, K., Bid, H. & Konwar, R. Reinventing the ACE inhibitors: some old and new implications of ACE inhibition. Hypertens Res 33, 11–21 (2010). https://doi.org/10.1038/hr.2009.184
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2009.184
Keywords
This article is cited by
-
The renin-angiotensin-aldosterone system (RAAS) signaling pathways and cancer: foes versus allies
Cancer Cell International (2023)
-
The bidirectional association between atrial fibrillation and myocardial infarction
Nature Reviews Cardiology (2023)
-
Subclinical cardiac damage in cancer patients before chemotherapy
Heart Failure Reviews (2022)
-
Purification of Angiotensin-Converting Enzyme (ACE) from Sheep Kidney and Inhibition Effect of Reduced Nicotinamide Adenine Dinucleotide (NADH) on Purified ACE Activity
Cell Biochemistry and Biophysics (2022)
-
Risk factors for thrombotic events in Korean patients with systemic lupus erythematosus
Scientific Reports (2021)
