
Abstract
Grape powdery mildew (PM), caused by the biotrophic ascomycete Erysiphe necator, is a devastating fungal disease that affects most Vitis vinifera cultivars. We have previously identified a panel of V. vinifera accessions from Central Asia with partial resistance to PM that possess a Ren1-like local haplotype. In this study, we show that in addition to the typical Ren1-associated late post-penetration resistance, these accessions display a range of different levels of disease development suggesting that alternative alleles or additional genes contribute to determining the outcome of the interaction with the pathogen. To identify potential Ren1-dependent transcriptional responses and functions associated with the different levels of resistance, we sequenced and analyzed the transcriptomes of these Central Asian accessions at two time points of PM infection. Transcriptomes were compared to identify constitutive differences and PM-inducible responses that may underlie their disease resistant phenotype. Responses to E. necator in all resistant accessions were characterized by an early up-regulation of 13 genes, most encoding putative defense functions, and a late down-regulation of 32 genes, enriched in transcriptional regulators and protein kinases. Potential Ren1-dependent responses included a hotspot of co-regulated genes on chromosome 18. We also identified 81 genes whose expression levels and dynamics correlated with the phenotypic differences between the most resistant accessions ‘Karadzhandahal’, DVIT3351.27, and O34-16 and the other genotypes. This study provides a first exploration of the functions associated with varying levels of partial resistance to PM in V. vinifera accessions that can be exploited as sources of genetic resistance in grape breeding programs.
Similar content being viewed by others


Introduction
The ascomycete Erysiphe necator can parasitize any green tissue of a grapevine causing a disease known as grape powdery mildew (PM, henceforth). Decline in leaf photosynthetic capacity1 and reduction in berry quality2 are typical symptoms of the disease. All commercial Vitis vinifera cultivars are susceptible to PM forcing grape growers to apply frequent and costly fungicide treatments throughout the growing season. In the European Union over 80 000 tons of fungicides are applied annually to control PM, which corresponds to about 67% of the total fungicides used for all crops, although only 3.3% of the agricultural land is dedicated to grape production.3 In California as much as 20% of the costs associated with wine grape cultivation are related to PM control.4 Excessive fungicide use is not only detrimental to the environment and human health but it also increases selection pressure on pathogen populations to adapt and acquire increasing levels of fungicide resistance.5
Erysiphe necator is considered to have originated in North America and is currently found in all grape-growing regions in the Northern and Southern hemisphere. It was introduced to Europe during the second half of the XIX century spreading throughout the continental and Mediterranean regions on susceptible V. vinifera cultivars. The genetic resistance often displayed by American grape species, of either Muscadinia or Vitis genera,6–10 supports the hypothesis of their co-evolution with E. necator populations. In M. rotundifolia major PM-resistant loci were mapped on both chromosome 12 (Run1)7 and 18 (Run2.1 and Run2.2).6 Despite the presumed American origin of the disease, resistance to PM has also been found in accessions of the Chinese grape species V. romanetii,6,11 V. piasezkii (M. A. Walker and S. Riaz, personal communication), and V. pseudoreticulata.12–14 Resistance to PM was also identified in Central Asian accessions of V. vinifera. A dominant PM resistance locus (Ren1) was mapped on chromosome 13 of the V. vinifera spp. sativa accessions ‘Kishmish vatkana’ and ‘Karadzhandal’ (syn.: ‘Dzhandzhal kara’).15,16 In ‘Kishmish vatkana’ Ren1 resistnce was shown to restrict hyphal growth and conidiation after successful penetration of epidermal cells, which indicates the involvement of late defense responses to E. necator.15 Further exploration of wild and cultivated V. vinifera germplasm from Central Asia identified 10 additional cultivated and wild accessions with partial resistance to PM, based on the presence of molecular markers known to be linked to Ren1 in ‘Kishmish vatkana’ and ‘Karadzhandal’.17 The newly identified accessions were presumed to carry a Ren1-like local haplotype based on the similar sequences of one of the co-segregating markers and identical allelic profiles with markers linked to the Ren1 locus.17 The presence of PM resistance in wild V. vinifera spp. sylvestris, the progenitor of the cultivated V. vinifera spp. sativa, indicates that PM resistance was potentially present in Central Asia for longer time than it has been earlier presumed and questions the origin of E. necator in North America.17 These accessions also displayed varying levels of resistance to PM in field trials,17 reflecting the wider genetic basis of PM resistance. This germplasm provides a unique opportunity to profile transcriptional changes that are induced by PM infection and gain insights into the complex multilayer defense strategy that grapevines from Central Asia have evolved against E. necator. The wider genetic basis and different horticultural attributes of this germplasm make it unique for breeding efforts, especially to stack resistance genes with different mechanisms to obtain more durable field resistance.
The type of reactions activated when specific resistance genes are expressed have been traditionally described using histochemical approaches.18 High-throughput methods that monitor genome-wide transcriptional changes can help further characterize at the molecular level the pathways regulated by defense-related genes. Genome-wide transcriptional profiling approaches have been effectively used to characterize disease resistance processes.14,19–24 In this study, we applied RNA sequencing approaches (RNAseq) to identify genes whose expression correlates with PM resistance in a panel of two wild and five cultivated Central Asian V. vinifera accessions. Through comparative analyses focused on differentially expressed genes shared across all the resistant genotypes but not the susceptible V. vinifera control, we identified functions that may contribute to the Ren1-dependent resistance mechanisms as well as gained insights into the molecular bases of the varying levels of resistance among these accessions. Grapevines were inoculated under controlled conditions with a highly virulent E. necator isolate and mock-inoculated controls were included. Disease susceptibility was assessed macro- and microscopically, and by quantitative PCR. Transcriptomes were sequenced at two time points after inoculation. Results are discussed in light of core inducible gene expression and their potential association with Ren1-dependent responses. Altogether the results showed that these Central Asian V. vinifera accessions activate distinct transcriptional responses to E. necator likely contributing to the partial resistance to powdery mildew.
Materials and methods
Plant material and assessment of PM susceptibility
Geographical origin, germplasm repository, and accession IDs of the grape genotypes used in this study are listed in Table 1. ‘Khalchili’, ‘Husseine’, and O34-16 were collected by Dr Harold P. Olmo in the 1950s, while ‘Sochal’ cuttings were imported into the USA in 1971 from the N. I. Vavilov Institute of Plant Industry in Saint Petersburg, Russia. ‘Late Vavilov’, a first-degree relative of ‘Karadzhandal’, is a V. vinifera spp. sativa variety of unclear origin with limited records in the Vitis International Variety Catalog (http://www.vivc.de) and the European Vitis database (http://www.eu-vitis.de). DVIT3351.27 was collected by Dr Ed Stover in Armenia. The French V. vinifera cv. ‘Carignan’ was included as a fully PM-susceptible positive control. PM disease evaluations were carried out using a detached leaf assay as described in Jones et al.5 The third and fourth fully expanded leaves were collected from multiple shoots of disease-free potted vines grown in a greenhouse. Leaves were surface-sterilized in 0.3% sodium hypochlorite solution. PM inoculations were carried out with a settling tower as described in Reifschneider and Boiteux.25 A settling tower (50×50×120 cm) attached to a vacuum system was used with 5 minutes of vacuum and 10 minutes of conidia settling time for all inoculations. Conidia of two week-old E. necator C-strain5 were used as PM inoculum (mean inoculum: ∼2.18 ± 1.5 conidia/mm2). PM growth was assessed by two approaches: (i) visual scoring of infection rate under a microscope and (ii) measurement of E. necator accumulation by quantitative PCR (qPCR) at four time points (1, 5, 10, and 14 days after inoculation, dpi; for details see below). Four biological replicates were used for each time point and the whole experiment was repeated three times. Visual assessment of disease severity was carried out by two people independently using a 1–5 scale: 1 = no hypha; 2 = five to ten germinated conidia per leaf with only primary hyphae; 3 = several germinated conidia with primary and, in some cases, secondary hyphae; 4 = mycelium is visible on the leaf surface with few conidiophores; 5 = mycelial development is severe with extensive conidiation clearly visible with the naked eye. For better visual observation, staining with Coomassie Brilliant Blue R-250 was carried out on detached leaves using the procedure described in Riaz et al.17
Erysiphe necator-infected material for RNAseq
Six plants of each accession were propagated from green cuttings and established in a greenhouse. Mock and PM inoculations were performed in two separate Conviron PGR15 growth chambers (UC Davis Controlled Environment Facility). For each accession three plants were mock-inoculated and three plants were PM-inoculated as described below. The two growth chambers were maintained at identical environmental conditions, which were set to favor E. necator infections at 60% humidity, 23°C during the day (16 hours), 15°C during the night. Pure E. necator isolate C-strain5 conidia were collected from detached leaf cultures, suspended in 0.001% Tween 20 solution and adjusted to a concentration of 5 × 105 conidia mL−1. Conidia were applied to the leaves using a portable paint sprayer (Perval). At the same time, grapevines in the other chamber were mock-inoculated with a 0.001% Tween solution. Leaves from each plant were collected at two time points (1 day and 5 dpi) at the same time of the day and immediately frozen in liquid nitrogen. Two PM- or mock-inoculated leaves from an individual plant were pooled and constituted a biological replicate. A total of three biological replications were obtained for each treatment. RNA was extracted using a Cetyltrimethyl Ammonium Bromide (CTAB) extraction protocol described in Blanco-Ulate et al.26 Two micrograms of total RNA were enriched for polyadenylated mRNA and used to prepare sequencing libraries following the Illumina TruSeq RNA Sample Preparation Kit v2. For quality control of size and purity, libraries were visualized on an Agilent Technologies 2100 Bioanalyzer using an Agilent High Sensitivity DNA chip.
RNAseq and read processing
cDNA libraries were sequenced in single-end mode with 50 cycles using an Illumina HiSeq2500 sequencer (DNA Technology core, UC Davis). A total of eight lanes were sequenced. Raw reads were deposited to the National Center for Biotechnology Information’s Gene Expression Omnibus (GEO) and are accessible through GEO (GSE67191 accession; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE67191). Sickle (v.1.2.1; https://github.com/ucdavis-bioinformatics/sickle) with a threshold of Q>20 and Scythe (v.0.991; https://github.com/vsbuffalo/scythe) with a prior of 0.4 were used to trim adapters and quality filter the reads, respectively. Reads were mapped onto a combined V. vinifera cv. ‘PN40024’ (version V1 from http://genomes.cribi.unipd.it/grape/) and E. necator C-strain5 reference using Bowtie2 v.2.2.327 with parameters: -q –end-to-end –sensitive –no-unal -p 20. Ambiguously mapped reads were excluded from further analysis using the program sam2counts.py (v0.9.1; https://github.com/vsbuffalo/sam2counts). Details on results of data trimming and mapping are reported in Supplementary Dataset S1.
Assessment of genetic diversity
The STAR28 2-pass mapping protocol was implemented for variant calling, because it was shown to improve alignment accuracy as well as sensitivity compared to other RNAseq mapping tools.29 PCR and optical duplicates were removed with Picard tools (v.1.119 http://broadinstitute.github.io/picard/). GATK (v.3.3.0) HaplotypeCaller was used to call sequence variants with parameters: -ploidy 2 -stand_call_conf 20.0 -stand_emit_conf 20.0 -dontUseSoftClippedBases. Homozygous SNPs were incorporated into synthetic transcripts for each accession using the GATK FastaAlternateReferenceMaker. The phylogenetic analysis was performed using the Hamming Distance implemented in the phyclust R package30 (v.0.1–15) based on the multiple alignment of all concatenated synthetic transcripts. PAML’s Yn00 (v.4.8) was implemented on reconstructed and original ‘PN40024’ transcripts to calculate ω (Supplementary Dataset S2). SnpEff31 (v.4.1a) was used along with V. vinifera gene annotations to call non-synonymous and synonymous substitutions. Unmapped reads were assembled using Trinity (v.2.0.6) with default parameters.32 TransDecoder (v.2.0.1; https://transdecoder.github.io) was used to extract the longest open reading frames from the de novo assembled transcripts. Predicted peptides matching known transposable elements associated proteins (RepBase33) were removed. ESTs were functionally annotated using Blast2GO.34
Calculation of differential expression
Read count normalization and significant differential expression were computed using DESeq2 v.2.1.2.10.35 Genes were considered constitutively down-regulated in the PM-partially resistant accessions if all seven varieties measured fold-changes (log2FC) ≤ –0.5 (P ≤ 0.05) relative to ‘Carignan’ at both 1 and 5 dpi, and considered constitutively over-expressed in the PM-partially resistant varieties if all seven measured log2FC ≥ 0.5 (P ≤ 0.05) at both 1 and 5 dpi (Supplementary Dataset S3). Log2FC in response to PM was computed with study-wide Deseq2 normalized counts plus an added pseudocount or set to ‘NA’ if a gene was not detected in both conditions (Supplementary Dataset S4; normalization factors are listed in Supplementary Dataset S1, raw counts can be downloaded from GEO http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE67191). Common PM-responsive genes associated with partial resistance were identified by applying a double fold-change cutoff: common up-regulated genes were genes with log2FC ≥ 0.5 in at least six Central Asian accessions and log2FC < 0.01 in ‘Carignan’; common down-regulated genes were genes with log2FC ≤ −0.5 in at least six Central Asian accessions and log2FC ≥ −0.01 in ‘Carignan’. Multiple factor analyses (MFA) were performed in R using the FactoMineR package (http://factominer.free.fr).
Gene Ontology (GO) term enrichments were computed using the classic Fisher method implemented in the topGO R package (v.2.18).36 GO term visualizations were created using the ‘Reduce + Visualize Gene Ontology’ (REViGO37) webserver (http://revigo.irb.hr) with resulting terms of topGO computations as input. REViGO sub-graphs were formed by selecting the SimRel algorithm38 and allowing for similarity values ranging from 0.7 to 0.9.
Quantitative PCR (qPCR)
Relative E. necator biomass accumulation was measured by qPCR. Grape and E. necator DNA were extracted using a modified CTAB protocol as described in Jones et al.5 from the same detached PM-infected leaves used for visual assessment of disease susceptibility. All DNA samples were randomized across reaction plates, and each plate had two replicates of uninfected V. vinifera and pure E. necator DNA samples as reference. E. necator elongation factor EnEF1 (KHJ34692) was amplified along with V. vinifera actin VvActin39 to quantify E. necator biomass as described in Jones et al.5
cDNA for reverse transcription qPCR (qRT-PCR) was prepared from total RNA as described in Blanco-Ulate et al.40 VvActin39 was used as a reference gene and processed in parallel with the grape target genes under evaluation. Primer sequences designed for this study are provided in Supplementary Table S1. The specificity of the primer sets was checked by analyzing the dissociation curves at temperatures ranging from 60 to 95°C. Three to four biological replicates of the infected and healthy material per accession were used to obtain the relative gene expression data. All qPCR reactions were carried out on a StepOnePlus PCR System using SYBR Green Master Mix (Applied Biosystems) and the following conditions: 95°C for 10 min, followed by 40 cycles of 95°C for 3 s and 60°C for 30 s. The formula 2(REFERENCE CT – TARGET CT) described in Chen and Dubcovsky41 was used to linearize the transcript levels for all genes.
Results
A panel of Central Asian grape accessions with varying levels of partial resistance against powdery mildew
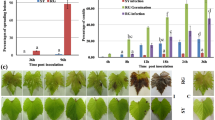
Two wild and five cultivated Central Asian V. vinifera accessions that display partial resistance against PM were the subject of this study (Table 1). To describe their disease phenotype, fungal growth and development of symptoms were assessed after leaf inoculation with the virulent E. necator C-strain. The fully susceptible V. vinifera cv. ‘Carignan’ was evaluated in parallel as a positive control. Conidia germination and establishment of primary appressoria were detected at 1 day post-inoculation (1 dpi) on leaves of all accessions. By 5 dpi, extensive growth and branching of fungal mycelia were observed in the susceptible ‘Carignan’ leaves, whereas in all PM-partially resistant accessions mycelial growth was restricted (Supplementary Figure S1). At 14 dpi (Figure 1a), categorical visual scores were assigned to describe disease development (Figure 1b) and correlated strongly with qPCR measurements of E. necator biomass (R2 = 0.89; P = 0.00041; Figure 1c). The greatest amount of fungal biomass and conidiophores was observed on the surface of ‘Carignan’ leaves (Figure 1a), in addition to widespread PM symptoms (i.e., white dusty appearance). Localized necrotic spots associated with appressoria, suggestive of a hypersensitive response and programmed cell death (PCD), were evident in the inoculated leaves of all PM-resistant accessions (Figure 1a). However, despite their overall similar partial resistance to the disease, the abundance of E. necator biomass and the presence of reproductive structures varied significantly across the Central Asian accessions at 14 dpi (Figure 1a). These results suggest that different mechanisms may contribute to the resistance phenotype in the different genotypes. ANOVA analysis followed by Tukey’s HSD post-hoc test (P ≤ 0.05) of both visual symptoms and biomass accumulation divided the seven Central Asian accessions into three groups (Figure 1b). Leaves of ‘Late Vavilov’ were the most infected among the Central Asian accessions, followed by ‘Husseine’, ‘Sochal’, and ‘Khalchili’. The lowest amount of E. necator growth and reproduction were present in leaves from ‘Karadzhandal’ and DVIT3351.27. Based on biomass accumulation O34-16 had similar level of resistance to the other V. vinifera spp. sativa DVIT3351.27 (Figure 1c).

Characterization of PM susceptibility in the eight V. vinifera cv (a) Micrographs of E. necator infection at 14 dpi. Bars = 0.2 mm (b) Mean visual scores of PM susceptibility in the eight cultivars. Different letters depict significant differences based on ANOVA followed by Tukey’s HSD test (P ≤ 0.05). (c) Correlation between visual scores of disease susceptibility and E. necator biomass measured by qPCR.
Sequencing and analysis of the transcriptomes of the seven Central Asian accessions and ‘Carignan’
RNAseq was performed to characterize the transcriptomes of the seven Central Asian accessions and the PM-susceptible cultivar ‘Carignan’. Sequencing libraries were prepared from leaf tissue collected at 1 and 5 dpi with E. necator C-strain,5 including mock-inoculated controls for each genotype and for each time point. Three biological replications, each represented by an individual plant, were included for each treatment. On average, 12 073 196 ± 1 890 923 raw reads per sample were obtained, of which 98.8 ± 1.0% were retained after quality trimming and filtering. A total of 87.6 ± 0.9% of the reads aligned to the combined reference of all 29 971 transcripts from V. vinifera ‘PN40024’42 and the 6533 transcripts predicted for E. necator C-strain.5 Only a small proportion of the total reads mapped on E. necator, ranging from 756 in ‘Karadzhandal’ to 67 857 in ‘Carignan’ at 5 dpi, which reflects the limited accumulation of E. necator biomass on the resistant accessions compared to the PM-susceptible control. Across all genotypes and conditions, the expression of 27 172 grape genes was detected.
Reads that did not map on the ‘PN40024’ genome scaffolds were de novo assembled to identify protein-coding genes that are present in the sequenced transcriptomes and absent in the reference genome. A total of 1430 ESTs derived from putative protein-coding genes were assembled. On average 178.9 ± 20.8 ESTs were obtained for each of the eight accessions, ranging from 148 in ‘Carignan’ to 199 and 209 in ‘Khalchili’ and ‘Husseine’, respectively. These de novo assembled transcripts represented functions potentially associated with disease responses including ethylene-responsive transcription factors, receptor-like kinases, and NBS-LRRs, as well as a large number of proteins associated with transposable elements. These results are in agreement with recent studies showing a remarkable structural variation between grape genomes.43,44
Transcript sequence analyses confirmed the high degree of heterozygosity in the grape genome.45–47 Similarly to what was previously reported by Velasco et al.,48 a heterozygous SNP rate of 2.30 ± 0.10 SNPs/Kb in the protein-coding regions was observed. This confirmed that rates of exonic heterozygous SNPs within an individual grape genotype are higher than the rates of fixed exonic SNPs between accessions (1.57 ± 0.08 SNPs/Kb). Homozygous SNP data were incorporated into reconstructed synthetic genes of each accession to enable further analysis of genetic diversity (Figure 2). Phylogenetic analysis based on multiple alignment of all reconstructed transcripts confirmed the closer genetic relatedness between first-degree relatives ‘Late Vavilov’ and ‘Karadzhandal’17 (group I) and between the two accessions originating from Afghanistan (‘Khalchili’ and ‘Husseine’, group II). Closer relationships between the two V. vinifera spp. sylvestris genotypes (O34-16 and DVIT3351.27, group III) and the susceptible varieties ‘Carignan’ and ‘PN40024’ (group IV) were also observed, while ‘Sochal’ did not cluster with any other accession.

Genetic diversity of the sequenced transcriptomes (a) Barplots showing the total number of homozygous SNPs resulting in synonymous or non-synonymous substitutions, or premature stop codons (b) Unrooted phylogenetic tree obtained from multiple alignment of synthetic transcripts reconstructed by incorporating the detected homozygous SNPs. Based on relatedness, genotypes were assigned to four groups.
Homozygous SNPs that result in non-synonymous amino acid changes or introduction of premature stop codons were analyzed to characterize the genetic diversity across accessions with potential functional relevance. An average of 25 418 ± 1321 non-synonymous substitutions relative to ‘PN40024’ were identified in each accession, ranging from 23 145 in ‘Carignan’ up to 27 893 in O34-16. This corresponded to 0.92 ± 0.05 non-synonymous substitutions per polymorphic gene (Figure 2a; Supplementary Dataset S2). An average of 105 ± 6 genes for each genotype carried SNPs that resulted in premature stop codons. These genes were further analyzed as they may represent potential functions involved in resistance or susceptibility to PM. Enrichment analysis of gene ontology (GO) terms (P < 0.05) revealed that PCD and defense responses are over-represented functions in genes with premature stop codons (Supplementary Dataset S2). Four genes with premature stops were identified in all seven resistant varieties, but not ‘Carignan’, these included: (i) a lysine histidine transporter 2 (VIT_01s0010g02640), (ii) a ribosomal protein L13 (VIT_14s0171g00250), (iii) a membrane bound O-acyltransferase (MBOAT; VIT_00s0507g00010), and (iv) a LRR receptor-like serine/threonine-protein kinase (VIT_00s0370g00010). The suppression of a lysine histidine transporter has been shown to confer broad-spectrum disease resistance in Arabidopsis.49,50
Rates of synonymous and non-synonymous substitutions (ω values or dN/dS ratios) were calculated by pairwise comparisons of the synthetic genes of each accession against the ‘PN40024’ reference genome. The analysis focused on the identification of genes with ω > 1, which indicates positive selection favoring amino acid replacements. In total 1971 (average per accession: 490.5 ± 35.8) genes had ω > 1 in the pairwise comparisons. These genes showed significant over-representation of 57 GO terms (P < 0.05), which included functions related to apoptotic processes and defense responses against pathogens (Figure 3; Supplementary Dataset S2). These results are in agreement with previous studies that reported elevated rates of non-synonymous over synonymous substitutions in genes associated with defense responses.51–57

GO term enrichment overlap between genes under positive selection (ω > 1), genes with higher constitutive expression in the Central Asian accessions, and genes with higher constitutive expression in ‘Carignan’ (a) Venn diagram showing numbers of overlapping and specific GO terms in each of the three groups of genes. (b) Graphical representation of relationships between enriched GO terms. To highlight closely related enriched GO terms, terms were organized in undirected sub-graphs using the REViGO web server.37 Colors indicate the gene group in which the GO terms are enriched, consistent with panel A. Dashed boxes indicate similar biological functions among the GO terms. Supplementary Dataset S3 provides all the GO terms and numeric IDs that are not described in panel C. (c) GO Categories with the greatest number of gene members are listed for each gene group, with the color coding of column 1 consistent with the gene groups shown in panel (a). Column 1 identifiers correspond to the circles in panel (b).
Identification of common constitutive transcriptional differences between PM-susceptible and PM-partially resistant grape accessions
Differences in constitutive expression of defense-related genes have been implicated in PM resistance of V. aestivalis ‘Norton’.22,58 Common functions constitutively differentially regulated in all Central Asian accessions compared to ‘Carignan’ could also potentially contribute to the partial resistance displayed by these varieties, although it cannot be ruled out that functions appear to be conserved because of closer genetic relatedness and geographical origins of the PM-tolerant accessions. To identify genes that are constitutively differentially expressed between all Central Asian accessions and the PM-susceptible ‘Carignan’, we compared gene expression between mock-inoculated ‘Carignan’ leaves and mock-inoculated leaves from each of the seven PM-partially resistant accessions. Forty-nine genes were consistently more expressed (log2FC ≥ 0.5, P ≤ 0.05) in the PM-partially resistant accessions when compared to ‘Carignan’, including a putative cinnamyl alcohol dehydrogenase (VIT_13s0047g00770) located between the closest flanking markers of the Ren1-locus.16 Sixty-three genes showed significantly higher expression levels in ‘Carignan’ when compared to the Central Asian accessions, including the lysine histidine transporter 2 (VIT_01s0010g02640) previously identified in this study to have a premature stop codon in all PM-partially resistant accessions (Supplementary Dataset S3). In Figure 3, we highlight the remarkable overlap between functions enriched in the set of genes constitutively differentially expressed, both up-regulated and down-regulated, and in the set of genes under positive selection (ω > 1). Worth noting is the shared group of GO terms associated with immune responses and cell death, which suggest that biotic stresses constitute an important component of the selection pressure exerted on grape populations.
A set of genes that represented constitutive differences among the PM-partially resistant accessions and ‘Carignan’ were validated by qRT-PCR using healthy leaves from a completely new set of plants grown under different environmental conditions (Supplementary Figure S2). The genes selected for validation belong to enriched biological functions that may be associated with either resistance or susceptibility to PM (e.g., plant immunity, cell death, hormone metabolism; Figure 3, Supplementary Table S1). The expressions of two genes encoding a receptor serine/threonine kinase (VIT_00s0425g00030) and a lipoxygenase (VIT_06s0004g01480) were validated to be constitutively higher in ‘Carignan’ than in the Central Asian accessions. The former gene may be involved in signaling cascades during pathogen recognition, while the latter is a key biosynthetic gene for the production of jasmonic acid, a hormone involved in defense responses against necrotrophs but not biotrophs.59 Among the genes with higher expression in the PM-resistant accessions, a multidrug and toxic compound extrusion gene (MATE; VIT_00s0225g00080) and a indole-3-butyric acid response 10 gene (IBR10; VIT_05s0049g00160) were validated to have higher expression levels in the PM-resistant accessions compared to ‘Carignan’. Both genes could play a role in PM resistance: the MATE transporter may function in the transport of antimicrobial compounds across the membrane, whereas the IBR10 may be part of the auxin signaling and responses to biotic stress. The BCL-2-associated athanogene 5 (VIT_01s0146g00150), which is involved in regulation of cell death, was confirmed to have a higher constitutive expression only in DVIT3351.27.
Identification of common transcriptional responses in the Central Asian accessions
To characterize transcriptional responses to E. necator, differentially expressed genes were identified by comparing mock-inoculated and PM-infected leaves from each accession. A union of 12 818 genes was differentially expressed in response to E. necator across all eight genotypes (P < 0.05). On average 776.1 ± 358.7 and 1541.8 ± 633.4 genes were significantly up-regulated in the seven resistant accessions at 1 and 5 dpi, respectively. E. necator also caused the down-regulation of 645 ± 344.7 genes at 1 dpi and 956.4 ± 535.2 at 5 dpi. A similar number of PM-responsive genes was detected in ‘Carignan’ at both time points. The largest number of PM-responsive genes at both time points was found in ‘Karadzhandal’, while ‘Late Vavilov’ showed the smallest number of differentially regulated genes in response to PM (Figure 4a and 4b).

Transcriptional responses to PM infection in the Central Asian accessions Barplots indicating the number of differentially regulated genes (P <0.05) at 1 (a) and 5 (b) dpi. Circos75 plots showing common differentially expressed genes between at least 6 resistant accessions at 1 (c) and 5 (d) dpi. Red and blue lines represent common up- and down-regulated genes, respectively. External bands correspond to each of the 19 chromosomes (+ Chr00) where the genes are located. A full list of genes represented by the connections in (c) and (d) are listed in Table 2. (e) Enriched GO terms (P <0.05) within the set of resistant-specific genes listed in Table 2. Darker colored circles represent terms with lower P values. Organization of sub-graphs is determined by REViGO web server.37 Boxes around terms group terms based on functional similarities, including (I) plant growth, (II) cell adhesion, (III) defense responses, (IV), cuticle formation, (V) phosphorylation, and (VI) cellular metabolism.
Differentially expressed PM-responsive genes in all accessions were compared to identify core transcriptional responses associated with partial PM resistance and potentially part of a Ren1-dependent resistance. In total, we found 51 genes differentially regulated in response to E. necator in at least six of the seven Central Asian accessions and that followed opposite trends or were not differentially regulated in the PM-susceptible ‘Carignan’ (Table 2). At 1 dpi, 13 genes were consistently up-regulated in the resistant accessions, while no other gene was found to be consistently down-regulated with the defined fold-change cutoffs (Figure 4c; Table 2). The 13 common differentially expressed genes at 1 dpi comprised an interesting set of inducible defense functions, including a pathogenesis related protein 1 precursor (PRP1; VIT_03s0088g00890), an ethylene-responsive transcription factor (ERF105; VIT_16s0013g00990), and five receptor kinases (VIT_00s2659g00010, VIT_04s0008g00310, VIT_09s0002g03020, VIT_16s0148g00030, VIT_19s0014g04130). At 5 dpi, only six genes were up-regulated in the PM-partially resistant accessions (Table 2), including two putative receptor serine/threonine-protein kinases (VIT_00s0258g00010, VIT_00s0294g00060) and a putative CC-NB-LRR (VIT_05s0102g01010). Notably, late common PM responses (5 dpi) in the Central Asian accessions involved a larger number of down-regulated genes (Table 2; Figure 4d). These 32 genes represent significant enrichment in nucleotide binding-related functions and protein kinase activities (P < 0.05; Figure 4e).
Chromosome 18 had the greatest number of differentially expressed genes in the PM-partially resistant varieties. Multiple PM resistance loci including Ren4 in V. romanetii, Run2.1, and Run2.2 in M. rotundifolia6 as well as 55% of the annotated NB-LRR genes are located on this chromosome.60 A positional gene enrichment analysis (PGE61) of chromosome 18 detected a significant over-representation of differentially regulated genes in response to PM between coordinates 3 401 893 to 8 421 561 (Figure 5), a region encompassing 496 protein-coding genes. Interestingly, the same region of chromosome 18 was enriched for genes undergoing positive selection (Figure 5d). Expression of the seven PM-responsive genes located in this region was confirmed by qRT-PCR (94.64% validation for 56 comparisons; Figure 5e).

Genomic region with significant enrichment in PM-responsive genes in the Central Asian accessions. (a) Heatmap showing gene density along chromosome 18. Colors range from dark blue (smaller gene density) to red (greater gene density). (b) Graphical representation of the PGE analysis results. Bars represent regions of chromosome 18 with significant enrichment in differentially expressed genes (P < 0.05). (c) Graphical representation of the location of protein-coding genes in the region identified by PGE analysis. Each box represents the exon of a protein-coding gene. (d) Locations of genes that are either detected as differentially expressed specifically in the Central Asian accessions in response to E. necator (blue) or constitutively more expressed in the Central Asian accessions than in ‘Carignan’. Gray boxes identify genes in the same regions that are under positive selection (ω > 1). (e) Correlation between PM-induced fold-changes measured by qRT-PCR and RNAseq.
PM-responsive genes specifically associated with the susceptible phenotype were also detected. Six genes at 1 dpi and 11 genes at 5 dpi were found to have significant differential expression only in ‘Carignan’. At 1 dpi, VIT_00s0187g00330, encoding a protein with a IQ calmodulin-binding domain similar to the apoptosis regulator Bcl-2 protein, was up-regulated in ‘Carignan’ and either down-regulated or not detected in all resistant varieties. In ‘Carignan’, E. necator caused the down-regulation of a guanine-methyltransferase (VIT_08s0040g01440) and a PHD transcription factor (VIT_04s0069g00140). At 5 dpi, ‘Carignan’-specific differential regulation included the down-regulation of an NBS-LRR receptor (VIT_07s0005g06240) and the up-regulation of genes encoding a S-receptor kinase 1 (VIT_11s0016g04890) and a BTB/POZ domain-containing protein (VIT_18s0122g01340).
Identification of transcriptional activities that correlate with the different levels of disease resistance
To visualize similarities in the PM responses among all eight varieties, principal coordinate analysis (PCoA) was performed with the pairwise intersections of the differentially expressed genes in response to E. necator (Supplementary Figure S3). Similarities in differentially expressed genes between accessions correlated more evidently with the pattern of disease susceptibility (Figure 1) than with genetic relatedness (Figure 2b). The first and second principal components of the PCoA for 5 dpi data clearly separated ‘Karadzhandal’, the most resistant of all Central Asian accessions from the most susceptible accessions, ‘Late Vavilov’, and ‘Carignan’ (Supplementary Figure S3b). The same pattern was evident when the PCoA was performed using enriched GO terms for each pairwise comparison between accessions (Supplementary Figure S4).
Multivariate analysis was performed to identify genes whose expression correlates with the most resistant disease phenotypes. Fold changes, along with PM visual scores and qPCR measurements of E. necator biomass, were used as quantitative variables in a multiple factor analysis (MFA, Figure 6a). The most resistant accessions ‘Karadzhandal’, O34-16, and DVIT3351.27 separated from the other Central Asian accessions along dimension 1, with ‘Late Vavilov’, the most susceptible genotype isolated from the rest. Similar clustering of accessions was obtained when normalized gene expression counts were used (Supplementary Figure S5). Variables with strong correlation with the main dimension 1 were extracted (R ≥ 0.7, P < 0.01) and filtered to identify genes specifically associated with the transcriptional profiles of the three most resistant accessions. Thirty-five PM-responsive genes were discovered with similar patterns of differential regulation in ‘Karadzhandal’, O34-16, and DVIT3351.27 and opposite trends in ‘Carignan’. We also identified 46 genes consistently more expressed in ‘Karadzhandal’, O34-16, and DVIT3351.27 compared to ‘Carignan’ (Supplementary Figure S5b). Functions related to hormone regulation, cellular transport, pectin metabolism, and xenobiotic catabolic processes were significantly over-represented in this set of 81 genes (Figure 6c). Interesting candidates included NB-LRRs (VIT_13s0139g00130, VIT_13s0158g00210, VIT_12s0034g01260, VIT_09s0002g04950), a cinnamyl alcohol dehydrogenase (VIT_00s0615g00010), and a MATE transporter (VIT_03s0038g00410) similar to EDS5, an essential component of salicylic acid signaling in Arabidopsis.62

Specific responses to PM among the most resistant Central Asian accessions (a) Multiple factor analysis (MFA) of the seven PM-partially resistant genotypes using three variables: log2 fold-changes of differentially expressed genes, PM visual scores, and E. necator biomass measured by qPCR (as previously reported in Figure 1). Each point represents a biological replicate for a given genotype. Ellipses define confidence areas (95%) for each genotype, while black squares represent their correspondent center of gravity. DIM, dimension. (b) Enriched GO terms (P < 0.05) in the set of genes that drive the separation of ‘Karadzhandal’, DVIT3351.27, and O34-16 in the MFA (R > 0.7, P < 0.01). To highlight closely related enriched GO terms, terms were organized into undirected sub-graphs using the REViGO web server.37 (c) Genes correlated with the most resistant accessions (positive dimension 1) in Panel (a), which also present a contrasting expression pattern in the susceptible ‘Carignan’ accession.
Discussion
Late post-penetration resistance is associated with partial resistance in the Central Asian accessions
In this study, we sequenced and analyzed the transcriptomes of seven Central Asian grape accessions that were previously shown to carry a Ren1-like local haplotype.17 Phylogenetic analyses using homozygous SNPs confirmed the genetic relationships reported in Riaz et al.17 The clustering reflected geographical origin and relatedness between modern cultivated varieties and sylvestris accessions. ‘Karadzhandal’ and ‘Late Vavilov’ have a first-degree parent progeny relationship with each other and were placed in the same clade. Similarly, two V. vinifera ssp. sylvestris accessions were closer to each other in comparison to cultivated V. vinifera spp. sativa accessions. We confirmed the partial resistance phenotype of these accessions, which we show is associated with a late post-penetration response to E. necator as previously observed in ‘Kishmish vatkana’15 and ‘Karadzhandal’.16 In all Central Asian accessions, E. necator germinated and established primary hyphae, but it failed to colonize extensively the leaf tissues. Differences between resistant and susceptible control plants became pronounced by day five, when the pathogen showed prolific production of mycelia and conidiophores on the PM-susceptible cv. ‘Carignan’, while it had limited growth and reproduction on resistant accessions. Necrotic spots that co-localized with appressoria of secondary hyphae suggest that late hypersensitive responses and PCD18,63 may be key in slowing down fungal spread in the Central Asian accessions as hypothesized in Hoffman et al.15
Comparative transcriptomics identifies common transcriptional activities potentially associated with Ren1 in the seven Central Asian accessions
Combinations of inducible and constitutive defenses may underlie the observed late penetration resistance phenotype. Transcriptome profiling revealed a set of 51 PM-responsive genes shared by all Central Asian accessions, which may be the signature of a common Ren1-dependent response (listed in Table 2). Interestingly, no common down-regulation was detectable at 1 dpi, while at 5 dpi most of the genes shared by the seven accessions were down-regulated, supporting the previous observation of sustained up-regulation of a larger number of defense-related genes during compatible interactions between a susceptible host and E. necator.22 Constitutive defenses were implicated in the resistance of V. aestivalis ‘Norton’ against PM.22,64 As a starting point to characterize potential constitutive defenses with resistance against PM, we identified a set of genes that had higher constitutive expression levels in the Central Asian accessions when compared to the susceptible cultivar ‘Carignan’ (listed in Supplementary Dataset S3). Interesting functions among these genes included detoxification of endogenous and exogenous secondary metabolites (i.e., MATE-efflux transporters), and auxin responses. In all Central Asian accessions, we also observed the constitutive up-regulation of VIT_13s0047g00770, a gene encoding a putative cinnamyl alcohol dehydrogenase (CAD) located between the closest markers flanking the Ren1 locus in ‘PN40024’.16 CAD gene expression was also reported to be constitutively higher in the PM-resistant V. aestivalis ‘Norton’ than in the susceptible ‘Cabernet Sauvignon’.58 CADs catalyze one of the final steps of the biosynthesis of monolignols and, thus, play a key role in tissue lignification.65 In wheat, disruption of CAD expression compromises papilla-associated cell wall reinforcement and penetration resistance to Blumeria graminis f.sp. titrici, the causal agent of wheat PM.66 CAD activity is also required for Mla1 resistance in barley, implicating this enzyme also in post-penetration HR responses.67
Different levels of PM resistance exist in the Central Asian germplasm
Despite the presence in all accessions of a Ren1-like haplotype and the similar late post-penetration resistance phenotype, we observed significant variation in the levels of PM infection among the Central Asian accessions. In this study, we identified 81 genes whose constitutive levels of expression or patterns of regulation in presence of E. necator correlate with the higher level of resistance displayed by O34-16, DVIT3351.27, and ‘Karadzhandal’. Further elucidation of the genetic bases of the different levels of resistance is only possible by direct sequence comparisons of the seven resistant haplotypes. Sequence analysis and further dissection by genetic mapping will resolve if the variation in disease susceptibility is due to different alleles of the same resistance gene resulting in different resistance specificities or to the unique combination of multiple genes. Analysis of the orthologous locus in ‘PN40024’ showed that the region is inherently prone to structural rearrangements and that NB-LRR genes proliferated through a combination of duplications and intragenic recombination events between paralogous genes.16 Similar genetic instability was found in the Mla PM resistance locus in barley.68 It is, thus, reasonable to hypothesize that among the Central Asian accessions there exists genetic variation within the Ren1-locus, as sequence polymorphisms, copy number variants, and presence/absence polymorphisms, which may underlie the different levels of resistance observed. Alternatively, epistatic interactions with genes outside of the Ren1-locus may be responsible for the distinct levels of resistance in the different genetic backgrounds.
Transcriptional profiling of gene families known to be involved in PM resistance
Specific genes have been previously implicated in grapevine resistance and susceptibility to PM (reviewed in Ref. 69), these include Enhanced Disease Susceptibility 1 (EDS1), Mildew Resistance Locus O (MLOs) genes, receptor-like kinases (RLKs), mitogen-activated protein kinases (MAPKs), WRKY transcription factors, and PR proteins. Transcriptional profiles of all gene families potentially implicated in PM responses based on published studies can be found in Supplementary Dataset S5, including the list of differentially regulated genes in other genome-wide studies.14,22,70,71 On average 61.0 ± 20.3% of the genes previously reported to be responsive to PM infections were found differentially expressed in our study (Supplementary Dataset S6). The incomplete overlap between transcriptional responses likely reflects not only differences in experimental conditions but also the diversity of resistance responses associated with the different Vitis genotypes. EDS1 has been shown to have high constitutive expression in PM-resistant V. aestivalis ‘Norton’ grapevines when compared to PM-susceptible V. vinifera ‘Cabernet Sauvignon’.72 In our study, at 1 dpi none of the ‘Carignan’ EDS genes were responsive to E. necator, whereas the EDS gene VIT_17s0000g07560 was up-regulated in five of the Central Asian accessions. MLOs have been shown to contribute to host susceptibility to PM by negatively regulating defense responses.73 Seventeen MLOs are annotated in the ‘PN40024’ genome and some have been hypothesized to be involved in PM susceptibility in V. vinifera based on their early transcriptional response to E. necator inoculation.70,74 Our results confirmed that VIT_13s0019g04060 (VvMLO770) was up-regulated in most grapevine accessions of this study, with the exception of DVIT3351.27. Remarkably, VIT_06s0004g03100 (VvMLO1370) was up-regulated at 5 dpi (log2FC = 0.93; P = 0.001) only in the PM-susceptible ‘Carignan’ accession, suggesting a role of this gene in PM susceptibility. None of these specific EDS and MLO genes were differentially regulated in response to PM in the Chinese species V. pseudoreticulata,14 further indication of the complexity of the mechanisms grapevines have evolved to resist PM.
Conclusions
In this study, we characterized genes potentially associated with late post-penetration resistance phenotype displayed by seven V. vinifera accessions from Central Asia that carry a common Ren1-like local haplotype. As the physical map of the Ren1 locus is resolved and reconstructed in all seven accessions, re-mapping of the RNAseq reads generated in this study will enable the profiling of candidate genes that may be responsible for the different levels of partial resistance observed. The identification of novel sources of genetic resistance among V. vinifera cultivars is particularly important from a breeding perspective. The introgression into elite cultivars from a V. vinifera background can reduce the rounds of backcrosses necessary to decrease the linkage drag, which could affect the distinctive characteristics of the cultivar that is the objective of genetic improvement. Stacking by marker-assisted selection of genes that confer partial resistance with single dominant resistance genes will help achieve more durable field resistance. Further genetic studies combined with comparative transcriptomic analyses with additional PM-resistant accessions from Asia (e.g., V. piasezkii and V. romanetii) and North America (e.g., M. rotundifolia) will define the different mechanisms underlying the resistance to PM available in the grape germplasm and guide breeding efforts to combine multiple resistance loci, while minimizing functional redundancies.
References
Nail WR, Howell GS . Effects of powdery mildew of grape on carbon assimilation mechanism of potted ‘Chardonnay’ grapevines. Hortscience 2004; 39: 1670–1673.
Stummer BE, Francis IL, Zanker T, Lattey KA, Scott ES . Effects of powdery mildew on the sensory properties and composition of Chardonnay juice and wine when grape sugar ripeness is standardised. Aust J Grape Wine R 2005; 11: 66–76.
Eurostat. The use of plant protection products in the European Union: data 1992–2003. Luxembourg: Office for Official Publications of the European Communities, 2007.
Fuller KB, Alston JM, Sambucci OS . The value of powdery mildew resistance in grapes: evidence from California. Wine Econ Policy 2014; 3: 90–107.
Jones L, Riaz S, Morales-Cruz A, Amrine KC, McGuire B et al. Adaptive genomic structural variation in the grape powdery mildew pathogen. Erysiphe necator. BMC 2014; 15: 1081.
Riaz S, Tenscher AC, Ramming DW, Walker MA . Using a limited mapping strategy to identify major QTLs for resistance to grapevine powdery mildew (Erysiphe necator) and their use in marker-assisted breeding. Theor Appl Genet 2011; 122: 1059–1073.
Barker CL, Donald T, Pauquet J, Ratnaparkhe MB, Bouquet A et al. Genetic and physical mapping of the grapevine powdery mildew resistance gene, Run1, using a bacterial artificial chromosome library. Theor Appl Genet 2005; 111: 370–377.
Dalbo MA, Ye GN, Weeden NF, Wilcox WF, Reisch BI . Marker-assisted selection for powdery mildew resistance in grapes. J Am Soc Hortic Sci 2001; 126: 83–89.
Fischer BM, Salakhutdinov I, Akkurt M, Eibach R, Edwards KJ et al. Quantitative trait locus analysis of fungal disease resistance factors on a molecular map of grapevine. Theor Appl Genet 2004; 108: 501–515.
Gadoury DM, Cadle-Davidson L, Wilcox WF, Dry IB, Seem RC, Milgroom MG . Grapevine powdery mildew (Erysiphe necator): a fascinating system for the study of the biology, ecology and epidemiology of an obligate biotroph. Mol Plant Pathol 2012; 13: 1–16.
Mahanil S, Ramming D, Cadle-Davidson M, Owens C, Garris A et al. Development of marker sets useful in the early selection of Ren4 powdery mildew resistance and seedlessness for table and raisin grape breeding. Theor Appl Genet 2012; 124: 23–33.
Wang Y, Liu Y, He P, Chen J, Lamikanra O, Lu J . Evaluation of foliar resistance to Uncinula necator in Chinese wild Vitis species. Vitis 1995; 34: 159–164.
Wan Y, Schwaninger H, Puchao H, Wang Y . Comparison of resistance to powdery mildew and downy mildew in Chinese wild grapes. Vitis 2007; 46: 132–136.
Weng K, Li Z-Q, Liu R-Q, Wang L, Wang Y-J, Xu Y . Transcriptome of Erysiphe necator-infected Vitis pseudoreticulata leaves provides insight into grapevine resistance to powdery mildew. Hort Res 2014; 1: 14049.
Hoffmann S, Di Gaspero G, Kovacs L, Howard S, Kiss E et al. Resistance to Erysiphe necator in the grapevine ‘Kishmish vatkana’ is controlled by a single locus through restriction of hyphal growth. Theor Appl Genet 2008; 116: 427–438.
Coleman C, Copetti D, Cipriani G, Hoffmann S, Kozma P et al. The powdery mildew resistance gene REN1 co-segregates with an NBS-LRR gene cluster in two Central Asian grapevines. BMC Genet 2009; 10: 89.
Riaz S, Boursiquot J, Dangl G, Lacombe T, Laucou V et al. Identification of mildew resistance in wild and cultivated Central Asian grape germplasm. BMC Plant Biol 2013; 13: 149.
Feechan A, Kabbara S, Dry IB . Mechanisms of powdery mildew resistance in the Vitaceae family. Mol Plant Pathol 2011; 12: 263–274.
Wise RP, Moscou MJ, Bogdanove AJ, Whitham SA . Transcript profiling in host-pathogen interactions. Annu Rev Phytopathol 2007; 45: 329–369.
Wang J, Yu H, Weng X, Xie W, Xu C et al. An expression quantitative trait loci-guided co-expression analysis for constructing regulatory network using a rice recombinant inbred line population. J Exp Bot 2014; 65: 1069–1079.
Tao YZ, Hardy A, Drenth J, Henzell RG, Franzmann BA et al. Identifications of two different mechanisms for sorghum midge resistance through QTL mapping. Theor Appl Genet 2003; 107: 116–122.
Fung RW, Gonzalo M, Fekete C, Kovacs LG, He Y et al. Powdery mildew induces defense-oriented reprogramming of the transcriptome in a susceptible but not in a resistant grapevine. Plant Physiol 2008; 146: 236–249.
Boddu J, Cho S, Kruger WM, Muehlbauer GJ . Transcriptome analysis of the barley-Fusarium graminearum interaction. Mol Plant Microbe Interact 2006; 19: 407–417.
Amrine KC, Blanco-Ulate B, Cantu D . Discovery of core biotic stress responsive genes in Arabidopsis by weighted gene co-expression network analysis. PLoS One 2015; 10: e0118731.
Reifschneider FJB, Boiteux LS . A vacuum-operated settling tower for inoculation of powdery mildew fungi. Phytopathol 1988; 78: 1463–1465.
Blanco-Ulate B, Vincenti E, Powell AL, Cantu D . Tomato transcriptome and mutant analyses suggest a role for plant stress hormones in the interaction between fruit and Botrytis cinerea. Front Plant Sci 2013; 4: 142.
Langmead B, Salzberg SL . Fast gapped-read alignment with Bowtie 2. Nat Methods 2012; 9: 357–359.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013; 29: 15–21.
Engstrom PG, Steijger T, Sipos B, Grant GR, Kahles A et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat Methods 2013; 10: 1185–1191.
Chen W . Overlapping codon model, phylogenetic clustering, and alternative partial expectation conditional maximization algorithm. Ames: Iowa State University, 2011.
Cingolani P, Platts A, Wang le L, Coon M, Nguyen T et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012; 6: 80–92.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 2011; 29: 644–652.
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J . Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 2005; 110: 462–467.
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M . Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005; 21: 3674–3676.
Love MI, Huber W, Anders S . Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15: 550.
Alexa A, Rahnenfuhrer J . topGo: Enrichment analysis for Gene Ontology. R package version 2.20.0, 2010.
Supek F, Bosnjak M, Skunca N, Smuc T . REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 2011; 6: e21800.
Schlicker A, Domingues FS, Rahnenfuhrer J, Lengauer T . A new measure for functional similarity of gene products based on Gene Ontology. BMC Bioinformatics 2006; 7: 302.
Licausi F, Giorgi FM, Zenoni S, Osti F, Pezzotti M, Perata P . Genomic and transcriptomic analysis of the AP2/ERF superfamily in Vitis vinifera. BMC Genomics 2010; 11: 719.
Blanco-Ulate B, Morales-Cruz A, Amrine KC, Labavitch JM, Powell AL, Cantu D . Genome-wide transcriptional profiling of Botrytis cinerea genes targeting plant cell walls during infections of different hosts. Front Plant Sci 2014; 5: 435.
Chen A, Dubcovsky J . Wheat TILLING mutants show that the vernalization gene VRN1 down-regulates the flowering repressor VRN2 in leaves but is not essential for flowering. PLoS Genet 2012; 8: e1003134.
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007; 449: 463–467.
Venturini L, Ferrarini A, Zenoni S, Tornielli GB, Fasoli M et al. De novo transcriptome characterization of Vitis vinifera cv. Corvina unveils varietal diversity. BMC Genomics 2013; 14: 41.
Da Silva C, Zamperin G, Ferrarini A, Minio A, Dal Molin A et al. The high polyphenol content of grapevine cultivar tannat berries is conferred primarily by genes that are not shared with the reference genome. Plant Cell 2013; 25: 4777–4788.
Sefc KM, Lopes MS, Mendonca D, Dos Santos MR, Da Camara Machado ML, Da Camara Machado A . Identification of microsatellite loci in olive (Olea europaea) and their characaterization in Italian and Iberian olive trees. Mol Ecol 2000; 9: 1171–1173.
Charlesworth D, Charlesworth B . Inbreeding depression and its evolutionary consequences. Annu Rev Ecol Syst 1987; 18: 237–268.
Barnaud A, Lacombe T, Doligez A . Linkage disequilibrium in cultivated grapevine, Vitis vinifera L. Theor Appl Genet 2006; 112: 708–716.
Velasco R, Zharkikh A, Troggio M, Cartwright DA, Cestaro A et al. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS One 2007; 2: e1326.
Liu G, Ji Y, Bhuiyan NH, Pilot G, Selvaraj G et al. Amino acid homeostasis modulates salicylic acid-associated redox status and defense responses in Arabidopsis. Plant Cell 2010; 22: 3845–3863.
Ditt RF, Kerr KF, de Figueiredo P, Delrow J, Comai L, Nester EW . The Arabidopsis thaliana transcriptome in response to Agrobacterium tumefaciens. Mol Plant Microbe Interact 2006; 19: 665–681.
Paape T, Bataillon T, Zhou P, JYKono T, Briskine R et al. Selection, genome-wide fitness effects and evolutionary rates in the model legume Medicago truncatula. Mol Ecol 2013; 22: 3525–3538.
Meyers BC, Shen KA, Rohani P, Gaut BS, Michelmore RW . Receptor-like genes in the major resistance locus of lettuce are subject to divergent selection. Plant Cell 1998; 10: 1833–1846.
Koenig D, Jimenez-Gomez JM, Kimura S, Fulop D, Chitwood DH et al. Comparative transcriptomics reveals patterns of selection in domesticated and wild tomato. P Natl Acad Sci USA 2013; 110: E2655–2662.
Di Genova A, Almeida AM, Munoz-Espinoza C, Vizoso P, Travisany D et al. Whole genome comparison between table and wine grapes reveals a comprehensive catalog of structural variants. BMC Plant Biol 2014; 14: 7.
Couch BC, Spangler R, Ramos C, May G . Pervasive purifying selection characterizes the evolution of I2 homologs. Mol Plant Microbe Interact 2006; 19: 288–303.
Buschiazzo E, Ritland C, Bohlmann J, Ritland K . Slow but not low: genomic comparisons reveal slower evolutionary rate and higher dN/dS in conifers compared to angiosperms. BMC Evol Biol 2012; 12: 8.
Bergelson J, Dwyer G, Emerson JJ . Models and data on plant-enemy coevolution. Annu Rev Genet 2001; 35: 469–499.
Fung RM, Qiu W, Su Y, Schachtman D, Huppert K et al. Gene expression variation in grapevine species Vitis vinifera L. and Vitis aestivalis Michx. Genet Resour Crop Ev 2007; 54: 1541–1553.
Glazebrook J . Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu Rev Phytopathol 2005; 43: 205–227.
Yang S, Zhang X, Yue JX, Tian D, Chen JQ . Recent duplications dominate NBS-encoding gene expansion in two woody species. Mol Genet Genomics 2008; 280: 187–198.
De Preter K, Barriot R, Speleman F, Vandesompele J, Moreau Y . Positional gene enrichment analysis of gene sets for high-resolution identification of overrepresented chromosomal regions. Nucleic Acids Res 2008; 36: e43–e43.
Nawrath C, Heck S, Parinthawong N, Metraux JP . EDS5, an essential component of salicylic acid-dependent signaling for disease resistance in Arabidopsis, is a member of the MATE transporter family. Plant Cell 2002; 14: 275–286.
Weng X, Wang L, Wang J, Hu Y, Du H et al. Grain number, plant height, and heading date7 is a central regulator of growth, development, and stress response. Plant Physiol 2014; 164: 735–747.
Fekete C, Fung RW, Szabo Z, Qiu W, Chang L et al. Up-regulated transcripts in a compatible powdery mildew-grapevine interaction. Plant Physiol Biochem 2009; 47: 732–738.
Voxeur A, Wang Y, Sibout R . Lignification: different mechanisms for a versatile polymer. Curr Opin Plant Biol 2015; 23: 83–90.
Bhuiyan NH, Selvaraj G, Wei Y, King J . Gene expression profiling and silencing reveal that monolignol biosynthesis plays a critical role in penetration defence in wheat against powdery mildew invasion. J Exp Bot 2009; 60: 509–521.
Zeyen RJ, Bushnell WR, Carver TLW, Robbins MP, Clark TA et al. Inhibiting phenylalanine ammonia lyase and cinnamyl-alcohol dehydrogenase suppresses Mla1 (HR) but not mlo5 (non-HR) barley powdery mildew resistances. Physiol Mol Plant P 1995; 47: 119–140.
Wei F, Wing RA, Wise RP . Genome dynamics and evolution of the Mla (powdery mildew) resistance locus in barley. Plant Cell 2002; 14: 1903–1917.
Qiu W, Feechan A, Dry I . Current understanding of grapevine defense mechanisms against the biotrophic fungus (Erysiphe necator), the causal agent of powdery mildew disease. Hort Res 2015; 2: 15020.
Winterhagen P, Howard SF, Qiu W, Kovács LG . Transcriptional up-regulation of grapevine MLO genes in response to powdery mildew infection. Am J Enol Viticult 2008; 59: 159–168.
Guo C, Guo R, Xu X, Gao M, Li X et al. Evolution and expression analysis of the grape (Vitis vinifera L.) WRKY gene family. J Exp Bot 2014; 65: 1513–1528.
Gao F, Shu X, Ali MB, Howard S, Li N et al. A functional EDS1 ortholog is differentially regulated in powdery mildew resistant and susceptible grapevines and complements an Arabidopsis eds1 mutant. Planta 2010; 231: 1037–1047.
Buschges R, Hollricher K, Panstruga R, Simons G, Wolter M et al. The barley Mlo gene: a novel control element of plant pathogen resistance. Cell 1997; 88: 695–705.
Feechan A, Jermakow AM, Torregrosa L, Panstruga R, Dry IB . Identification of grapevine MLO gene candidates involved in susceptibility to powdery mildew. Funct Plant Biol 2008; 35: 1255–1266.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R et al. Circos: an information aesthetic for comparative genomics. Genome Res 2009; 19: 1639–1645.
Acknowledgements
This work was supported by funding to Dario Cantu from the American Vineyard Foundation (Grant: 2013–1510) and from the College of Agricultural and Environmental Sciences (UC Davis) and from the Louis P. Martini Endowment to M. Andrew Walker. Laura Jones was supported by the Adolf L. & Richie C. Heck Research Fellowship, the Frank H. Bartholomew Scholarship, the John Ferrington Award, the Robert L. Balzer Scholarship, the Southeast Pennsylvania Region of the American Wine Society Scholarship, the Stewart Good Scholarship, the Wine Spectator Scholarship, and the Women for WineSense Scholarship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Supplementary information accompanies the article can be found on the Horticulture Research’s website (http://www.nature.com/hortres).
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Amrine, K., Blanco-Ulate, B., Riaz, S. et al. Comparative transcriptomics of Central Asian Vitis vinifera accessions reveals distinct defense strategies against powdery mildew. Hortic Res 2, 15037 (2015). https://doi.org/10.1038/hortres.2015.37
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hortres.2015.37
This article is cited by
-
Identification of powdery mildew resistance in wild grapevine (Vitis vinifera subsp. sylvestris Gmel Hegi) from Croatia and Bosnia and Herzegovina
Scientific Reports (2022)
-
Construction of a high-density genetic map and detection of a major QTL of resistance to powdery mildew (Erysiphe necator Sch.) in Caucasian grapes (Vitis vinifera L.)
BMC Plant Biology (2021)
-
VviERF6Ls: an expanded clade in Vitis responds transcriptionally to abiotic and biotic stresses and berry development
BMC Genomics (2020)
-
Identification of SNPs and InDels associated with berry size in table grapes integrating genetic and transcriptomic approaches
BMC Plant Biology (2020)
-
Genetic mapping and survey of powdery mildew resistance in the wild Central Asian ancestor of cultivated grapevines in Central Asia
Horticulture Research (2020)
