
Abstract
Peach (Prunus persica) and almond (Prunus dulcis) are two sexually compatible species that produce fertile offspring. Almond, a highly polymorphic species, is a potential source of new genes for peach that has a strongly eroded gene pool. Here we describe the genetics of a male sterile phenotype that segregated in two almond (‘Texas’) × peach (‘Earlygold’) progenies: an F2 (T×E) and a backcross one (T1E) to the ‘Earlygold’ parent. High-density maps were developed using a 9k peach SNP chip and 135 simple-sequence repeats. Three highly syntenic and collinear maps were obtained: one for the F2 (T×E) and two for the backcross, T1E (for the hybrid) and E (for ‘Earlygold’). A major reduction of recombination was observed in the interspecific maps (T×E and T1E) compared to the intraspecific parent (E). The E map also had extensive monomorphic genomic regions suggesting the presence of large DNA fragments identical by descent. Our data for the male sterility character were consistent with the existence of cytoplasmic male sterility, where individuals having the almond cytoplasm required the almond allele in at least one of two independent restorer genes, Rf1 and Rf2, to be fertile. The restorer genes were located in a 3.4 Mbp fragment of linkage group 2 (Rf1) and 1.4 Mbp of linkage group 6 (Rf2). Both fragments contained several genes coding for pentatricopeptide proteins, demonstrated to be responsible for restoring fertility in other species. The implications of these results for using almond as a source of novel variability in peach are discussed.
Similar content being viewed by others


Introduction
Cytoplasmic male sterility (CMS) is driven by rearrangements in the mitochondrial genome that result in plants being unable to produce fertile pollen.1 Usually, CMS is a binary system where the products of one or more nuclear genes (restorer genes) interfere with the causal mitochondrial proteins and reestablish fertility.1,2 CMS is widespread in the plant kingdom and has been described in more than 150 species2 including members of the Poaceae, Leguminosae, Umbelliferae, Brassicaceae, Chenopodiaceae, Solanaceae, and Liliaceae. Various cases of male sterility have been reported in the Rosaceae, as in Fragaria vesca,3 peach,4,5 Japanese apricot,6 almond,7 and pear.8 Of these, CMS has only been documented for peach, Japanese apricot and pear. CMS is an important model to analyze the interplay between organelle and nuclear genomes that has implications in the evolution of sex in plants,9 and is a key technological aspect in the development of F1 hybrid seed for many of the major herbaceous crops.10
Peach is the only species of cultivated Prunus (which also includes cherry, almond, plum, and apricot) that does not have a functional gametophytic self-incompatibility system and behaves as self-pollinating. This character, along with its important economic value, have made peach a model species for genetic studies of Prunus, and many major genes have been described and mapped on its genome.11,12 Peach is also one of the best characterized species of the Rosaceae family and its whole genome sequence has recently been published.13
Self-pollination is the major factor that explains the low level of genetic diversity of peach.13,14,15 In contrast, the almond genome is highly variable, as has been revealed with molecular markers,16 and may be a source of novel alleles that could confer new properties and provide raw materials for characters such as disease resistance, extended fruit shelf life, and organoleptic fruit quality in peach. These major objectives of peach breeding are difficult to achieve due to the scarcity of variability in its gene pool.
The fertility of the F1 and successive generations is one of the key elements of the success of interspecific crosses. F1 hybrids between almond and peach are usually fertile, but we found male sterile plants in their F2 and backcross one (BC1) progenies. Our data indicate that almond cytoplasm confers male sterility to the peach unless the almond allele of at least one of two independent restorer genes (Rf1 and Rf2) is present. These results have important implications for the process of introgression of almond genes into the peach background and thus for peach breeding.
Materials and methods
Plant material
We used the 80 individuals of the F2 progeny of the T×E mapping population, obtained by selfing the F1 hybrid plant ‘MB 1.37’ (almond ‘Texas’ × peach ‘Earlygold’), for which a high-density map already exists.11 We have recently added 31 plants to obtain a final mapping population of N = 111, improving its resolution. The original T×E population is kept at the IRTA Center of Cabrils (Barcelona, Spain) and a copy has been planted in the IRTA Experimental Station of Lleida at Gimenells (Spain), both grafted on ‘Garnem’ rootstocks. Additionally we developed a new BC1 population derived from the cross between the hybrid ‘MB 1.37’ and the peach ‘Earlygold’ of N = 190 that was named T1E. Original trees of T1E were planted on their own roots at Cabrils, and replicates were grafted on ‘Garnem’ rootstocks and planted in the field at Gimenells.
Phenotyping
The male sterility character was studied over three seasons (2010–2013) by visual inspection. The plants analyzed were those that were alive at the moment of phenotyping (90 for T×E and 174 for T1E). Male sterile plants were characterized by the lack of pollen and empty, white anthers, in contrast to the fertile plants which had pollen and anthers ranging from yellow to anthocyanic (Figure 1). In addition, the pollen germination capacity in the 121 T1E individuals that produced pollen was evaluated in vitro according the protocol described by17 with an additional 15% sucrose. At the end of the process, each plant was characterized either as sterile or fertile.

Male fertile and sterile phenotypes in the almond × peach F2 and BC1 progenies. (a) Parental flowers: ‘Texas’ (T), MB1.37 (F1), ‘Earlygold’ (E). (b) Flowers from four F2 individuals. (c) Flowers from four BC1 individuals. The first individual from each population (56 of T×E and 23 of T1E) were male sterile with white anthers and absence of pollen. The other individuals with colored anthers were fertile.
Marker detection, linkage map construction, and genetic analysis
Genomic DNA was extracted from young leaves using the CTAB method,18 omitting the final RNAse treatment step. Sample DNA quality and concentration were checked and measured with a DNA spectrophotometer (NanoDrop Technologies, Wilmington, USA).
Genetic maps were obtained using a set of 135 selected simple-sequence repeat (SSR) markers with good genome coverage, most of them previously located on the T×E map11,19 or of known genome physical position (Table S1). Four of these markers were developed based on the Genome Database for Rosaceae (GDR; http://www.rosaceae.org/) list of SSRs, to cover the distal regions of linkage groups two and six (G2 and G6). They were named CPP (C for CRAG, PP for Prunus persica) followed by a number coinciding with that of the SSR annotated at the IGA peach genome browser (http://services.appliedgenomics.org/fgb2/iga/prunus_public/gbrowse/prunus_public/). Their main characteristics are shown in Table S2. These markers were studied in the whole T1E population and in the 31 new T×E individuals. Most of the SSRs used were common between T×E and T1E (Table S1). Two additional SSRs (TPScp10 and TPScp10) developed from Japanese plum chloroplast20 were used to evaluate the almond or peach origin of the cytoplasm in the T1E population.
In addition, we genotyped 50 individuals of the T×E population and 123 individuals of T1E with the 9k Illumina Infinium SNP chip developed by the Peach SNP International Consortium.21 Genotyping was done at the Fondazione Edmund Mach (San Michele all’Adige, Italy) with DNA (50 ng/μl) extracted with the Qiagen DNAeasy 96 Plant Kit, as described before.21 Genotypes were scored with the GenomeStudio Data Analysis software (Illumina Inc.) using Gencall threshold of 0.15. Markers with a GeneTrain score <0.6 were excluded from the dataset used for mapping. SNPs with skewed segregations (P < 0.05) were used for mapping and discarded only if unlinked.
Linkage maps were constructed using all data available from markers (111 plants for T×E and 190 plants for T1E) with MapMaker/exp 3.022 with the Kosambi mapping function at a logarithm of odds (LOD) grouping threshold ≥3.0. All marker data for SSRs and SNPs were produced as part of this research with the exception of the 114 SSRs used in T×E that were mapped before using all11 or a subset19 of the 80 initial individuals of this population (see Table S1). Linkage maps were drawn using the MapChart 2.1 software.23 For the T1E progeny we obtained two maps, each with the data from one of the parents: the ‘Texas’ × ‘Earlygold’ F1 hybrid used as female parent (T1E map), and the map from the pollen donor ‘Earlygold’, called the E map. Linkage group terminology was as usual in Prunus.11,13
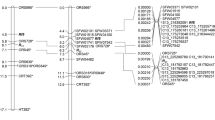
Given the high number of SNPs identified, we mapped only one per genome position or bin, i.e. group of markers with the same genotype for all individuals and separated by at least one recombination event from neighboring bins. A single SNP was used to label each bin, normally containing more markers. The selected marker was the closest to the physical origin of the bin that had all or most data for the individuals studied (i.e., excluding SNPs that were scored as dominant or heterozygous for the hybrid individual and ‘Earlygold’ in the T1E and E maps). The exception was 54 markers of the E map in which the SNPs used to label a specific bin were selected among those in common with T×E and T1E to facilitate visualization of the map comparison presented in Figure 2. The SSRs were all added as markers in the maps so they could be used as anchor points between the three maps studied and with published Prunus maps.

Linkage maps obtained, one with the almond (‘Texas’) × peach (‘Earlygold’) F2 population (T×E), and two with the BC1 population to peach, one for the hybrid female parent (T1E) and the other for the peach ‘Earlygold’ male parent I. Each marker position corresponds to a bin (i.e. a group of markers with the same genotype for all the plants studied, represented here by a single marker). Some of the anchor markers of each map are connected with lines. These selected anchor markers are all the SSRs and some of the SNPs, particularly of the E map, to show the completeness of the comparison between T1E and E. The names of the SNP markers are shortened to facilitate visualization of the maps. The two restorer genes are highlighted in red.
As results were inconclusive when the male sterility character was mapped as a single gene in the T×E, T1E, and E maps, we followed a two-step procedure. The data were first mapped as quantitative characters using MapQTL,24 using values of 2 for fertile, 1 for sterile and missing data for plants that could not be phenotyped. QTLs were identified with the interval mapping approach using the BC1 population type, and were declared significant when reaching a LOD > 3.0. Then, knowing the approximate positions of the two QTLs detected, we used the genotype data of these two loci to manually find their position in the full dataset of ordered markers of the two linkage groups where these loci were located. A unique and fully consistent position was found for both loci in the positions identified by MapQTL.
Candidate gene analysis
In CMS, fertility restorer (Rf) genes usually encode proteins of the pentatricopeptide (PPR) family.25 For this reason we explored the positions of the PPR genes in the Prunus genome that were identified using the information of annotated genes in its sequence (v1.0) (http://www.rosaceae.org/search/genes), using ‘pentatricopeptide protein’ as a keyword. We also checked for homologs of other non-PPR Rf genes of known sequence in the regions where the restorer genes found in this paper were located. This was done using tblastx on the GDR webpage, using the default options. The tested genes were Rf2 from maize,26 Rf2, Rf3, Rf4, and Rf17 from rice,27,28,29 and Rf1 from sugar beet.30
Results
Map construction and comparison
Three maps were obtained, T×E with 1948 markers (114 SSRs and 1834 SNPs), T1E, the BC1 female parent, with 2032 markers (113 SSRs and 1919 SNPs) and E, the peach male parent, with 1091 markers (1050 SNPs and 40 SSRs) identifying each of them the expected eight (G1–G8) linkage groups (Table 1; Figure 2).
The E map had approximately half of the markers that were polymorphic in the T×E and T1E. In contrast with the other two maps, where marker distribution was generally uniform, the distribution of markers in the E map was extremely heterogeneous. Chromosomal fragments had densities similar to T1E and T×E and others were without markers, resulting in large gaps (nine greater than 10 cM, and two in G2 and G6 longer than 25 cM), and vast distal regions of the genome had no segregating markers. The overall physical coverage was 155.4 Mbp compared to the 207.3 Mbp of T1E (75.0%) and the 211.9 Mbp of T×E (73.3%), and there was low coverage between mapped markers at the extremes of certain linkage groups (compared to T1E): coverage of G4, G5, and G8 was only 32.4%, 36.4%, and 70.2%, respectively of the distance of the same groups in T1E. Gaps were rare in T×E and T1E, four in total for each map with a maximum gap of 2.9 Mbp. However, there were 23 gaps of >2 Mbp in E, with a maximum gap of 19.7 Mbp in G4, adding to a total distance of 103.0 Mbp, almost half of the distance covered by T×E or T1E.
There were 95 SSRs in common between T×E and T1E, all in the same map position and order, except for CPPCT029 and CPPCT053 which were in a different order. T1E and E shared 40 SSRs, and 31 of these were also anchor points with T×E. A large proportion of the 1834 SNPs mapped in T×E were also mapped in T1E (97.5%) and vice versa (92.9%) for the 1919 SNPs mapped in T1E. These proportions were much lower in E, where only 478 SNPs (25.9%) were common with T×E and 491 (25.6%) with T1E. The order of the SNPs in T×E and T1E was identical, with SNP_IGA_155433 being the only exception, mapping to a slightly different position in G2 of both maps.
Discrepancies were found when linkage and physical maps were compared. The most relevant were two large inversions at the distal end of G1 (>5.1 Mbp), and the proximal end of G7 (>4.2 Mbp), misplacements of a >1.2 Mbp fragment of G2 at the proximal end of this group, a fragment of >2.2 Mbp of G4 mapped to the central part of G2, and another fragment of G4 (>0.4 Mbp), located in the central region of G6. Markers of the unmapped scaffolds S9, S10, S12, and S17 could be placed at a map position on G3, G3, G2, and G6, respectively. These data are in agreement with some of the sequence refinements introduced to the v1.0 version of the peach genome that can be found at the GDR (http://www.rosaceae.org/species/prunus_persica/genome_v1.0_refinements).
Genetic analysis of the male sterility character
Both chloroplast SSRs (TPScp10 and TPScp10) had bands of different sizes for ‘Texas’ and ‘Earlygold’. The MB1.37 hybrid and all the T×E and T1E progenies had the ‘Texas’ allele, as expected considering that ‘Texas’ and MB1.37 were used as the female parents.
Field observations on male sterility were fully consistent between years in the T×E and T1E populations. Pollen was viable in all individuals that produced it as indicated with the in vitro test. In T1E, of the 174 plants phenotyped, 121 were fertile and 53 male sterile, and in T×E, only one plant was male sterile whereas all the others that could be phenotyped (89) produced fertile pollen. We compared these segregations with those expected considering that this character was determined by two dominant epistatic loci (Rf1 and Rf2, for restorer of male fertility), where the almond parent carried the two dominant alleles Rf1 and Rf2, the peach parent was homozygous for the recessive alleles (rf1 and rf2), being their hybrid (MB1.37) heterozygous Rf1rf1/Rf2rf2. Only the individuals of T×E and T1E carrying the double recessive homozygote were sterile and the rest were fertile. Data from T1E were in agreement with the expected 3:1 segregation ratio (χ2 = 2.82; ns), but those of T×E significantly differed from a 15:1 (χ2 = 4.06; P = 0.04).
By QTL analysis of the male sterility character in the largest T1E population, two of its map regions were identified as harboring clear QTLs, while none were found in the E map. The two loci were in the proximal regions of G2 and G6 and were detected with LODs of 16.7 and 17.4, respectively, explaining 36% and 37% of the phenotypic variability, and 67% when considered together. Based on the positions detected by MapQTL, the two genes in G2 and G6 were manually located and found to have a unique position in these groups. Rf1 cosegregated with a bin at 3.8 cM from the top of G2 (SNP_IGA_144919 and 119 additional SNPs covering a physical distance of 3.4 Mbp), flanked by SNP_IGA_144913 (1,118,746 bp) and SNP_IGA_192890 (4,535,916 bp) at 3.0 and 4.6 cM, respectively, corresponding to a 3.4 Mbp total physical distance. Rf2 co-mapped in G6 with two bins containing the SNP_IGA_623894 and SNP_IGA_625843 markers (map positions 9.2 and 9.9 cM) and four more SNPs that spanned a distance of 0.7 Mbp, and was flanked by SNP_IGA_622231 (5,606,752 bp) and SSR UDP96-001 (7,040,897 bp) with map positions of 7.2 and 10.6 cM, respectively, and covering a total physical distance of 1.4 Mbp.
Of the 90 T×E progeny phenotyped, only the male sterile plant had a marker genotype compatible with the positions of Rf1 and Rf2 as mapped in T1E. Nevertheless, the position of these two loci could only be roughly estimated in T×E, corresponding to the 15.3 cM (∼14.3 Mbp) region between the bins defined by SNP_IGA_230270 and SNP_IGA_260361 in G2, for Rf1, and the 39.3 cM (∼15.5 Mbp) fragment between markers SNP_IGA_609630 and SSR pchgms5 for Rf2 in G6.
Search for candidate Rf genes
We found a total of 554 genes with PPR motifs in the peach genome sequence, four located in the 3.4 Mbp sequence of G2 containing Rf1 and 16 in the 1.4 Mbp of G6 encompassing Rf2 (Table S3). None of the six non-PPR Rf genes of known sequences that were tested had sequences with high similarity in the target regions. Only one gene highly homologous to the rice Rf17, which corresponds to an acyl-carrier protein synthase29 was located near the almond Rf2 region (macromolecule 6:5462574; e-value 2e-44).
Discussion
High-density maps were developed using two interspecific almond × peach populations with the 9k IPGI Illumina Infinium chip and a set of 135 SSRs, most of them of known position in the Prunus reference linkage map. The three maps constructed, one for the F2 (T×E) and two for the BC1 progeny (T1E and E) demonstrated the high quality of the data obtained in the following aspects: (1) as expected, the majority (>92%) of the markers were common to the T×E and T1E maps, as they are derived from the same individual (the hybrid plant MB1.37); (2) anchor markers between maps were syntenic and collinear with almost no exception; (3) the E map had a much higher level of recombination per unit of physical distance than the T×E and T1E maps, suggesting that the recombination rates in intraspecific progenies are higher than in interspecific, as observed in other interspecific crosses in Prunus31 and other plant species32; (4) the number of markers in the interspecific progenies for the SNP chip were around 2000 in both T×E and T1E, whereas only half segregated in the map of the peach intraspecific parent. The same 9k IPGI chip was used in the peach × peach progeny (‘Bolero’ × ‘OroA’)33 and also found fewer segregating markers (1450 for ‘Bolero’ and 350 for ‘OroA’). Moreover, the T×E and T1E maps had a homogeneous coverage of the whole genome, with only a few gaps >2 Mbp, none larger than 2.9 Mbp. Aside from the physical coverage of the maps usually being lower in the intraspecific maps, these results suggest either that ascertainment bias was not an important factor in the almond/peach materials or that it was compensated for by a higher level of polymorphism compared to peach materials alone, giving a similar or higher marker number and coverage. These results also indicate that the peach SNP chip can be used in other almond/peach progenies.
The map constructed with the peach ‘Earlygold’ was characterized by fragments with high marker density followed by regions without markers, in contrast with the almond × peach maps that had a relatively homogeneous distribution of markers. Similar results have been observed when using high-density SNP maps in other peach × peach crosses.33,34 These results indicate that the regions without markers may correspond to genome fragments that are identical by descent. These fragments may account for a large proportion of the genome, estimated in E as 103.0 Mbp (50% of the total physical distance covered by T1E). This hypothesis is supported by the recent history of cultivated peach, with most commercial European and North American cultivars coming from a bottleneck in the early US breeding programs, leading among other things to a high level of inbreeding.35,36 Moreover, the number of generations between the founders and the current commercial cultivars is probably very low considering the long life, propagation by grafting and breeding schemes (cultivars are usually selected from F1 progenies between two partly heterozygous parents) typical of fruit trees. This would lead to the maintenance of large chromosomal fragments, as supported by the high conservation of linkage disequilibrium of peach.15 The consequences are that the parents used by breeders are often close relatives, with large regions of their genomes and those of the cultivars resulting from their offspring being identical by descent, as our results suggest. This has implications for genome analysis and breeding, as only the parts of the genome that are heterozygous will segregate and only the crossovers produced at these heterozygous fragments will result in changes that may produce innovative gene combinations. This also means that only a part of the genome needs to be monitored when using markers for whole genome selection. Therefore maps with only partial marker coverage would be suitable for genetic analysis, provided that the parents are previously tested for a large initial sample of markers with good coverage of the genome, allowing identification of the segregating regions of each particular parent or cross.
Our results suggest that male fertility in T×E and T1E individuals, all with the almond cytoplasm, is determined by two independent almond restorer genes, Rf1 and Rf2. Presence of the dominant (almond) allele of either gene would result in a fertile plant. This hypothesis is confirmed by the observed data giving a 3:1 segregation for T1E and by the identification of two genome regions of 3.4 and 1.4 Mbp located in G2 and G6, respectively, where these two genes are located. Considering only nuclear inheritance, this hypothesis conflicts with the fertile ‘Earlygold’ phenotype because the absence of almond alleles at Rf1 and Rf2 would have implied sterility. Other simple hypotheses of Mendelian inheritance (one or two genes) involving only genetic male sterility were discarded by our data, as they imply that the ‘Earlygold’ parent should carry alleles conferring fertility in at least one locus. This locus should then segregate in the T1E population and be mapped in the E map, which did not occur.
The situation in T×E fitted well with the described two-locus model, although in this case the expected 15:1 segregation was not in agreement with the 89:1 segregation observed. This departure can be explained as the marker segregations had a lower frequency of homozygous peach alleles in both regions, significant in G6 (χ2 = 10.45; P = 0.001) but not in G2 (χ2 = 3.68; P = 0.06), indicating that they were selected against the peach alleles, as also noted previously.37
CMS is determined by the interaction between the mitochondrial and nuclear genomes.1,2 Our results are compatible with this model, where the products of two almond genes interact with an unknown mitochondrial-encoded protein of this species to determine the fertile phenotype. In peach these products do not exist, so fertility can only be recovered when the almond alleles are present. The most common nuclear factors involved in fertility restoration are PPR repeat proteins, ubiquitous in plant genomes.25 This also occurs in peach, where 554 genes containing PPR motifs were identified, some in the target regions of G2 (4) and G6 (16) where the Rf1 and Rf2 genes reside. These regions did not contain sequences with high homology to other known non-PPR restorer genes.25 The numbers of PPR candidates found in G2 were much lower than those in G6 for a much shorter region of the genome (3.4 vs. 1.4 Mbp). This may be due to the G2 region containing Rf1 being a possible centromeric region, as identified previously,13 with low gene density and low recombination, whereas Rf2 is located at the distal region of chromosome 6, with higher gene density and recombination. In G6, six of the PPR genes cluster in a narrow 87 kb region (ppa026767, ppa023798m, ppa019799m, ppa023651m, ppa023796m, and ppa015333m) and two pairs are in tandem (ppa026767/ppa023798m and ppa023796m/ppa015333m). Given that Rf genes that encode for PPR proteins are usually in clusters with other non-Rf PPR encoding genes,38 the PPR genes of this region and particularly those that are in tandem are the strongest candidates to be the cause of Rf2.
Male sterility, cytoplasmic or genetic, has been used as an efficient system for commercial production of F1 hybrid seed in herbaceous species.10 This is not a varietal type currently used in peach or most fruit tree crops as grafting is an efficient propagation procedure that allows multiplication of selected vigorous heterozygotes. However, certain male sterile cultivars such as ‘Chinese Cling’ and ‘JH Hale’39 are among the founders of the first US peach breeding programs. This was because of their outstanding performance as parents for breeding purposes36,40 and probably because all seeds collected from them come from crosses with neighboring plants, facilitating the usually tedious pollination procedure. The pollen sterile character was found to be determined by a single gene (Ps/ps),4 mapped at position 0.0 cM of G6.41 The closest marker on the physical map, the RFLP FG215 (position 8.8 cM on the map and 285,872 bp on the genome sequence) is not compatible with the position of Rf2 that we have placed in the 5,606,752–7,040,897 interval, suggesting that Ps and Rf2 are different loci. A second male sterility gene ps2 has been reported,5 although its map position is unknown. A case of male sterility for almond has been reported7 where a cross between the male sterile cv. Rof and a fertile peach heterozygous for Ps yielded only fertile hybrids, suggesting that, if male sterility was of nuclear origin, the gene or genes involved were other than Ps.
The presence of CMS has been reported in peach5 from accession PI 240928, although no evidence of restorer genes has been provided. The male sterility of certain accessions of Japanese apricot (Prunus mume) has also been shown to be produced by CMS, but in this case certain crosses with fertile individuals restored fertility, suggesting the presence of Rf alleles.6 Assuming that the CMS of ‘Texas’ can be generalized for almond, the consequences of this finding are that the introgression of genes from almond into peach would result in sterile individuals unless one of the Rf genes was also introgressed, or if the peach parent is used as the female donor in one of the crosses. This may also occur in progeny between peach and other closely related Prunus species. This has to be taken into account when planning crossing schemes to integrate new genes from these species into peach, so that the peach cytoplasm is recovered at some point in the process to avoid undesirable sterility problems.
Data archiving
Original data are included in Supplementary Tables S1, S2, and S3. Data used to construct the maps and phenotypic data are available at Genome Database for Rosaceae (http://www.rosaceae.org/).
References
Pelletier G, Budar F . The molecular biology of cytoplasmically inherited male sterility and prospects for its engineering. Curr Opin Biotechnol 2007; 18: 121–125.
Schnable PS, Wise RP . The molecular basis of cytoplasmic male sterility and fertility restoration. Trends Plant Sci 1998; 3: 175–180.
Tennessen JA, Govindarajulu R, Liston A et al. Targeted sequence capture provides insight into genome structure and genetics of male sterility in a gynodioecious diploid strawberry, Fragaria vesca ssp. bracteata (Rosaceae). G3 (Bethesda) 2013; 3: 1341–1351.
Scott DH, Weinberger JH . Inheritance of pollen sterility in some peach varieties. Proc Amer Soc Hort Sci 1938; 45: 229–232.
Werner DJ, Creller MA . Genetic studies in peach: inheritance of sweet kernel and male sterility. J Amer Soc Hort Sci 1997; 122: 215–217.
Yaegaki H, Miyake M . Haji T et al. Inheritance of male sterility in Japanese apricot (Prunus mume). HortSci 2003; 38: 1422–1423.
Alonso JM, Socias R . Lack of male-sterility allelism between peach and almond. Acta Hort 2003; 622: 257–260.
Thompson JM, Vanderwet T, Draper AD et al. Evidence of cytoplasmic and genetic male-sterility in pears. J Hered 1976; 67: 339–346.
Caruso CM, Case AL, Bailey MF . The evolutionary ecology of cytonuclear interactions in angiosperms. Trends Plant Sci 2012; 17: 638–643.
Kempe K, Gils M . Pollination control technologies for hybrid breeding. Mol Breed 2011; 27: 417–437.
Dirlewanger E, Graziano E, Joobeur T et al. Comparative mapping and marker assisted selection in Rosaceae fruit crops. Proc Natl Acad Sci USA 2004; 101: 9891–9896.
Arús P, Verde I, Sosinski B et al. The peach genome. Tree Genet Genomes 2012; 8: 531–547.
Verde I, Abbott AG, Scalabrin S et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nature Genet 2013; 45: 487–494.
Aranzana MJ, Illa E, Howad W et al. A first insight into peach [Prunus persica (L.) Batsch] SNP variability. Tree Genet Genomes 2012; 8: 1359–1369.
Li XW, Meng XQ, Jia HJ et al. Peach genetic resources: diversity, population structure and linkage disequilibrium. BMC Genet 2013; 14: 84.
LessMnejja M, Garcia-Mas J, Audergon JM et al. Prunus microsatellite marker transferability across rosaceous crops. Tree Genet Genomes 2010; 6: 689–700.
Asma BM . Determination of pollen viability, germination ratios and morphology of eight apricot genotypes. African J Biotechnol 2008; 7: 4269–4273.
Doyle JJ, Doyle JL . Isolation of plant DNA from fresh tissue. Focus 1990; 12: 13–15.
Howad W, Yamamoto T, Dirlewanger E et al. Mapping with a few plants: using selective mapping for microsatellite saturation of the Prunus reference map. Genetics 2005; 171: 1305–1309.
Ohta S, Nishitani C, Yamamoto T . Chloroplast microsatellites in Prunus, Rosaceae. Mol Ecol Notes 2005; 5: 837–840.
Verde I, Bassil N, Scalabrin S et al. Development and evaluation of a 9K SNP array for peach by internationally coordinated SNP detection and validation in breeding germplasm. PLoS One 2012; 7: e35668.
Lander ES, Green P, Abrahamson J et al. MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural population. Genomics 1987; 1: 174–181.
Voorrips RE . MapChart: Software for the graphical presentation of linkage maps and QTLs. J Hered 2002; 93: 77–78.
Van Ooijen JW, Boer MP, Jansen RC et al. MapQTL® 4.0 software for the calculation of QTL positions on genetic maps. Wageningen: Plant Research International, 2002.
Chen L, Liu YG . Male sterility and fertility restoration in crops. Ann Rev Plant Biol 2014; 65: 579–606.
Cui X, Wise RP, Schnable PS . The rf2 nuclear restorer gene of male-sterile T-cytoplasm maize. Science 1996; 272: 1334–1336.
Itabashi E, Iwata N, Fujii S et al. The fertility restorer gene, Rf2, for Lead Rice-type cytoplasmic male sterility of rice encodes a mitochondrial glycine-rich protein. Plant J 2011; 65: 359–367.
Luo D, Xu H, Liu Z et al. A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nat Genet 2013; 45: 573–577.
Fujii S, Toriyama K . Suppressed expression of retrograde-regulated male sterility restores pollen fertility in cytoplasmic male sterile rice plants. Proc Natl Acad Sci USA 2009; 106: 9513–9518.
Matsuhira H, Kagami H, Kurata M et al. Unusual and typical features of a novel restorer-of-fertility gene of sugar beet (Beta vulgaris L.). Genetics 2012; 192: 1347–1358.
Arús P, Yamamoto T, Dirlewanger E et al. Synteny in the Rosaceae. In: Jules Janick (ed.) Plant breeding reviews. Vol. 27. Hoboken, NJ: John Wiley and sons, 2005: 175–211.
Tanksley SD, Ganal MW, Prince JP et al. High density molecular linkage maps of the tomato and potato genomes. Genetics 1992; 132: 1141–1160.
Eduardo I,, Pirona R et al. Genetic dissection of aroma volatile compounds from the essential oil of peach fruit: QTL analysis and identification of candidate genes using dense SNP maps. Tree Genet Genomes 2013; 9: 189–204.
Martínez-García PJ, Parfitt DE, Ogundiwin EA et al. High density SNP mapping and QTL analysis for fruit quality characteristics in peach (Prunus persica L.). Tree Genet Genomes 2013; 9: 19–36.
Hesse CO . Peaches. In: Janick J Moore J N (eds.) Advances in fruit breeding. West Lafayette, IN: Purdue University Press, 1975: 285–335.
Scorza R, Mehlenbacher SA, Lightner GW . Inbreeding and coancestry of freestone peach cultivars of the eastern United States and implications for peach germplasm improvement. J Amer Soc Hort Sci 1985; 110: 547–552.
Joobeur T, Viruel MA, de Vicente MC et al. Construction of a saturated linkage map in Prunus using an almond × peach F2 progeny. Theor Appl Genet 1998; 97: 1034–1041.
Fujii S, Bond CS, Small D . Selection patterns on restorer-like genes reveal a conflict between nuclear and mitochondrial genomes throughout angiosperm evolution. Proc Natl Acad Sci USA 2011; 108: 1723–1728.
Okie WR . Handbook of peach and nectarine varieties. Agricultural Research Service. Byron, GA: Southeastern Fruit and Tree Nut Research Lab, 1998.
Weinberger JH . Characteristics of the progeny of certain peach varieties. Proc Amer Soc Hort Sci 1944; 45: 233–238.
Dirlewanger E, Cosson P, Boudehri K et al. Development of a second-generation genetic linkage map for peach [Prunus persica (L.) Batsch] and characterization of morphological traits affecting flower and fruit. Tree Genetics Genomes 2006; 3: 1–13.
Acknowledgements
This work has been partly funded under grants AGL2012-40228-C02-01 from the Spanish Ministry of Economy and Knowledge and the EU Seventh Framework Programme by the FruitBreedomics project No. 265582: Integrated approach for increasing breeding efficiency in fruit tree crops. The views expressed in this work are the sole responsibility of the authors and do not necessarily reflect the views of the European Commission. The authors wish to thank D. Micheletti and M. Troggio from Fondazione Edmund Mach (San Michele all’Adige, Italy) for help in the genotyping of the SNP chip and C. Fontich and A. Ortigosa (IRTA) for maintenance of the plant populations in Lleida and Cabrils, respectively.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplemental Information for this article can be found on the Horticulture Research website (http://www.nature.com/hortres).
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Donoso, J., Eduardo, I., Picañol, R. et al. High-density mapping suggests cytoplasmic male sterility with two restorer genes in almond × peach progenies. Hortic Res 2, 15016 (2015). https://doi.org/10.1038/hortres.2015.16
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/hortres.2015.16
This article is cited by
-
Inheritance and QTL analysis of chilling and heat requirements for flowering in an interspecific almond x peach (Texas x Earlygold) F2 population
Euphytica (2020)
-
Developing a methodology for identifying brown rot resistance in stone fruit
European Journal of Plant Pathology (2019)
-
Identification of a new allele of the Dw gene causing brachytic dwarfing in peach
BMC Research Notes (2018)
-
Mapping a major gene for red skin color suppression (highlighter) in peach
Euphytica (2017)
-
Genetic analysis of the slow-melting flesh character in peach
Tree Genetics & Genomes (2017)
