
Abstract
The apple (Malus × domestica Borkh.) cultivar ‘Su Shuai’ exhibits greater disease resistance, shorter internodes and lighter fruit flavor compared with its parents ‘Golden Delicious’ and ‘Indo’. To obtain a comprehensive overview of the sequence variation in these three horticultural traits, the genomes of ‘Su Shuai’ and ‘Indo’ were resequenced using next-generation sequencing and compared to the genome of ‘Golden Delicious’. A wide range of genetic variations were detected, including 2 454 406 and 18 749 349 single nucleotide polymorphism (SNP) and 59 547 and 50 143 structural variants (SVs) in the ‘Indo’ and ‘Su Shuai’ genomes, respectively. Among the SVs in ‘Su Shuai’, 17 genes related to disease resistance, 10 genes related to Gibberellin (GA) and 19 genes associated with fruit flavor were identified. The expression patterns of eight of the SV genes were examined using reverse transcription-quantitative polymerase chain reaction (RT-qPCR). The results of this study illustrate the genomic variation in these cultivars and provide evidence for a genetic basis for the horticultural traits of disease resistance, short internodes and lighter flavor exhibited in these cultivars. These results provide a genetic basis for the phenotypic characteristics of ‘Su Shuai’ and, as such, these SVs could serve as gene-specific molecular markers in maker-assisted breeding of apples.
Similar content being viewed by others


Introduction
Next-generation sequencing technology, an efficient and economic method of obtaining comprehensive sequence data, has become the primary tool for acquiring information on genetic variation within a species and identifying unique genotypic molecular markers. These capabilities allow a fuller characterization of genomes at the chromosome level, particularly for genes responsible for important agronomic traits.1,2 This approach, which has been successfully applied to human genomics through the ENCODE project,3 has similar potential for the genetic analysis and improvement of crop species.4
The discovery and quantification of genomic variants enables researchers to characterize genomic differences among specific genotypes. Genomic variants include changes in nucleotides and changes in chromosome structures. For trait mapping, nucleotide variants, such as single nucleotide polymorphisms (SNPs), are commonly used to characterize genotypic diversity. Insertions and deletions (InDels) are commonly used to investigate evolutionary divergence and speciation. Genomic (i.e., chromosomal) rearrangements longer than 50 nucleotides are often considered structural variants (SVs),5 because they have a direct effect on the structure and behavior of the chromosome and cause variation in gene dosage. SVs are the result of rearrangements within a chromosome or between chromosomes. The types of variation associated with SVs include insertions longer than 50 bp, inversions, duplications, translocations, mobile elements in the target genome (where they have been characterized) or a combination of such events.6
Genetic diversity resides mainly in genomic structural variants such as SNPs, short-sequence InDels, and inter- and intrachromosomal translocations and inversions.7 Therefore, it is likely that differences in phenotypic traits observed among ‘Golden Delicious’, ‘Indo’ and ‘Su Shuai’ cultivars of apple (Malus × domestica) are because of SNPs and SVs. Analyses of SNPs and InDels are commonly used in genetic and genomic studies such as the construction of genetic linkage maps and the identification of quantitative trait loci (QTL). In human studies, SVs have increasingly been considered a major driving force in evolution.8 SVs have been associated with important phenotypic traits, including several rare and complex diseases in humans.6 In plants, the associations between SVs and phenotypes have been less thoroughly studied, except in maize9 where SV comparisons have been analyzed among inbred lines10 and between maize and its progenitor, teosinte (Zea mays ssp. parviglumis H.H.Iltis & Doebley).11 Recent studies have also demonstrated SVs to be reflected by changes in copy number variation in Arabidopsis12 and intracultivar variation in soybean [Glycine max (L.) Merr.].13,14 Genomic tools also have tremendous potential in assisting Rosaceae (which includes pome fruits, stone fruits, strawberries and roses) crop breeders to map traits more precisely and efficiently, resulting in the development of new cultivars that are beneficial to both consumers and growers.15
Most fruit tree crops, such as the Malus species, are propagated vegetatively, which allows individual genetic composition to be maintained, including chromosomal variants that may play important roles in cultivar-specific phenotypic traits. Apple is a main fruit crop in temperate regions of the world and the fourth most economically important fruit crop worldwide, following citrus, grape and banana.16 Genomic resources for apple have been developed over the past 10 years, including the sequencing of the ‘Golden Delicious’ genome.17 The apple genome sequence anchored to a high-density linkage map has provided the apple research community with new tools to identify genes and other functional elements that will facilitate marker-assisted breeding and our understanding of plant genome structure. Transcriptomic, proteomic and metabolomic studies are also greatly benefiting from the availability of an annotated genome.17,18
The apple cultivar ‘Su Shuai’ was derived from a cross made in 1976 between ‘Golden Delicious,’ used as male parent, and ‘Indo,’ used as female parent (Figure 1).19 Compared with ‘Golden Delicious’, the surface of ‘Su Shuai’ is smooth and yellow green without any rust coloration. ‘Su Shuai’ was produced from an advanced breeding line (18-8) that was selected in 1987 after bearing fruits for three consecutive years. This cultivar exhibited stable traits over several years of continuous observation and was approved by the Jiangsu Province Crop Variety Approval Committee in China and named ‘Su Shuai’ in 2011. The main characteristics of ‘Su Shuai’ are high yields, short internodes, high resistance to diseases such as Alternaria blotch (Alternaria alternata), and a smooth fruit surface with no rust coloration.20,21 To obtain a comprehensive overview of sequence variations in ‘Su Shuai’, we resequenced the genome of ‘Su Shuai’ and its female parent ‘Indo’.

Pedigree of ‘Su Shuai’. ‘Su Shuai’ represents a cross between ‘Indo’ (female parent) and ‘Golden Delicious’ (male parent). The parents of ‘Indo’ are unknown, but may be a mutant or seedlings of ‘White winter Pearmain’. The parents of ‘Golden Delicious’ are also unknown, but are theorized to be ‘Golden Reinette’ × ‘Grimes Golden’.
Materials and methods
Plant material
Whole-genome resequencing was performed on two individuals from the apple cultivars ‘Indo’ and ‘Su Shuai’. The resulting sequences were aligned and compared to the ‘Golden Delicious’ reference genome and one another. The scions were grown in the experimental orchard of the Institute of Pomology, Chinese Academy of Agricultural Sciences, Xingcheng, China.
‘Su Shuai’ is a new apple cultivar derived from a cross between ‘Golden Delicious’ (male parent) and ‘Indo’ (female parent). The growth of ‘Su Shuai’ trees is robust and characterized by a compact canopy. The appearance of the fruit is truncate conical, with a smooth and green-yellow skin with small dots and an absence of fruit russeting. Fruit development occurs over 155 days. The average single fruit weight is 241 g and the average total yield is 45 t hm−2.19 In comparison to its parents, this cultivar exhibits strong disease resistance, particularly to Alternaria blotch (Alternaria alternata), and has short internodes and a light fruit flavor. The scions were obtained from an orchard of Fengxian County, Xuzhou City, Jiangsu Province, China.
Sequencing
Genomic DNA was extracted from leaves of two samples and treated with RNase I as described by Tong et al.,22 and then sequenced on an Illumina HiSeq2000 (Biomarker Technologies, Beijing, China) following Illumina protocols, producing a paired-end and a mate-pair library for each individual. The sequences were aligned to the ‘Golden Delicious’ apple reference genome (http://www.rosaceae.org/species/malus/malus_x_domestica/genome_v1.0p) using the Burrows-Wheeler Aligner software.23,24 The raw data were submitted to the SRA (Sequence Read Archive) (http://www.ncbi.nlm.nih.gov/sra/) and the corresponding accession number is SRP043674.
The binary alignment mapped files generated by the Burrows-Wheeler Aligner software25 were used to generate Simple Alignment Maps. Subsequently, variant calling filter files were generated using the mpileup routine in the software SAMtools.26 All statistical analyses were performed using SPSS 18.0 software.
SNP detection
SNPs were identified using SAMtools.23 The variation effect software SNPEFF3.3f (version 3.3f (2013) snpEff: variant effect prediction. http://snpeff.sourceforge.net/) was used to annotate the effects of the variations (e.g., synonymous or non-synonymous). A SnpEff predictor database file in binary format (.bin)27 was created to locate each SNP within annotated transcripts or intronic regions. Both HTML and text output files were generated from SnpEff 3.3f. The output included the position of the SNP on the scaffold, the reference nucleotide, the changed nucleotide, whether it was a transition or a transversion, transitions/transversions (Ts/Tv), warnings, gene ID, gene name, biotype, transcript ID, exon ID, exon rank effect, amino-acid change (old aa/new aa), old codon/new codon, codon number (based on the coding sequence (CDS)) and CDS size. Warnings were provided if the predicted changes differed from those predicted using the ‘apple v1.0p’ genome. Because a complete, reliable functional annotation for apple has not been completed, 5′ and 3′ UTR regions are not currently available for gene models.
SV detection
Pindel28 and BreakDancer29 software were used for the detection of structural variants. These programs are specifically designed to identify genomic structural variations through sliding window and clustering strategies by processing sorted binary alignment mapped or SAM files resulting from the alignment of sequences from ‘Indo’ and ‘Su Shuai’ to the ‘Golden Delicious’ reference genome.
Functional annotation and screening of differential genes
The sequences of the differential genes that SNPs or SV variations were located in were obtained. Differential genes were annotated with BLAST against the M. domestica reference genes (http://www.rosaceae.org/species/malus/malus_x_domestica) and Arabidopsis proteins (http://www.arabidopsis.org/). The annotation libraries are provided in Supplementary Table S1 using a threshold e-value of 10−5. To analyze main biological functions, we mapped all differential genes to the terms in the KEGG (Kyoto Encyclopedia of Genes and Genomes)30 (http://www.genome.jp/kegg/). Additionally, Pfam (protein families)31 (http://pfam.xfam.org/) annotation was performed to assign differential genes to different gene families.
Annotation information on homologous genes was used to screen for differential genes with horticultural traits. Non-synonymous SNPs or SVs were compared based on the phenotypes of ‘Su Shuai’ and its parent plants, and differential genes were identified with three horticultural traits in ‘Su Shuai’.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
RT-qPCR was performed to detect the relative change in expression of genes identified in the resequencing analysis. Total RNA samples from ‘Golden Delicious’, ‘Indo’, and ‘Su Shuai’ were isolated as described by Tong et al.,22 and used for expression analysis.
Total RNA (1 µg) from each sample was treated with DNase I (invitrogen, Carlsbad, California, United States) and used for cDNA synthesis. The first-strand cDNA synthesis was performed with an Oligo (dT) primer using SuperScript III RT (Invitrogen). The cDNA was diluted to 100 µg µL−1 and used for RT-qPCR reactions in 96-well plates in a 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, California, United States). Gene-specific primers for the identified genes were designed using the Beacon designer program. Each RT-qPCR reaction (20 µL) contained 8.6 µL water, 0.4 µL (100 nM) of forward and reverse primers, 10 µL of SYBR Green II Master and 1 µL of diluted cDNA. To normalize the total amount of cDNA in each reaction, a housekeeping gene, apple tubulin, was used in the control reactions. All reactions used the primers provided in Table 10. Relative gene expression levels were calculated using the 2−ΔCTΔCT method.
Results
Characterization of three horticultural traits in ‘Su Shuai’ and ‘Golden Delicious’
In this study, only ‘Su Shuai’ and ‘Golden Delicious’ were used for field experiments because ‘Indo’ apples were not planted in the orchard. Analysis of data from field experiments and phenotypic observations indicated that ‘Su Shuai’ exhibited greater variability in several quantitative traits compared with the variation observed in ‘Golden Delicious’. Three horticultural traits were evaluated: internode length, fruit flavor and disease resistance. A t-test was used to determine whether these traits were statistically significantly different in the two cultivars (‘Su Shuai’ and ‘Golden Delicious’). Internode length and soluble solid content exhibited a significantly greater variance (p<0.01) in ‘Su Shuai’ (2.37 cm average for internode length; 10.0% average for soluble solid content) compared with ‘Golden Delicious’ (3.49 cm, 12% for internode length and soluble solids content, respectively). The level of disease resistance exhibited little year-to-year difference in the field experiments. The resistance of ‘Golden Delicious’ and ‘Su Shuai’ to certain diseases, particularly A. alternata (apple pathotype), was tested using standard protocols for plant disease resistance. ‘Su Shuai’ was regarded as highly resistant (HR), whereas ‘Golden Delicious’ was categorized as moderately resistant over two successive years (Table 1).
Resequencing of ‘Su Shuai’ and ‘Indo’
Genomic variants among the three genotypes included both small variants (SNPs) and larger SVs, such as InDels. ‘Indo’, the female parent of ‘Su Shuai’, is more heterozygous than ‘Su Shuai’ when both are compared to ‘Golden Delicious’ (the genome reference sequence). A pair-wise comparison of consensus genome sequences indicated that ‘Indo’ was more divergent than ‘Su Shuai’ from ‘Golden Delicious’.
Of the 25.73 gigabases (Gb) of raw sequence data, approximately 10 Gb per individual remained after filtering the paired-end sequences of each cultivar (S2), which translated to an average of 23× depth of coverage per genotype, and the average insert length was 326 bp for each genotype. The genome sequence of ‘Gold Delicious’ available from the Genome Database for the Rosaceae (http://www.rosaceae.org) was used as a reference for the alignment. Each sample read sequence was compared to the reference genome using Burrows-Wheeler Aligner. The efficiency for aligning sequences from ‘Su Shuai’ and ‘Indo’ to the reference genome was approximately 80%, and the extent of coverage of the apple genome was greater than 96% (S3).
SNP detection
SNPs were the most common variant present in the two genotypes. SAMtools was used to detect SNPs. By filtering out questionable bases based on low quality and depth, a highly reliable mismatch (base mismatch) locus set was obtained. A total of 2 454 406 SNPs were detected in ‘Indo’, in which transitions accounted for 68.27% and transversions accounted for 31.73% of the identified SNPs. Approximately 79.09% were heterozygous and 20.91% were homozygous. A total of 1 874 349 SNPs were obtained for ‘Su Shuai’, with transitions accounting for 68.02% and transversions accounting for 31.98% of the SNPs. Approximately 89.82% were heterozygous and 20.91% were homozygous (Table 2). The distributions of SNP-types were similar among the 17 chromosomes in both cultivars (Table 3 and Figure 2). Most of the SNPs were intergenic: 1 219 463 (49.7%) in ‘Indo’ and 925 862 (49.4%) in ‘Su Shuai’. Many of the SNPs were located in the protein-coding regions of genes: 258,383 (10.5%) in ‘Indo’ and 214 398 (11.3%) in ‘Su Shuai’ (Table 4).

Diagrammatic representation of structural variation in the sequence data obtained from the resequencing of ‘Indo’ (a) and ‘Su Shuai’ (b). The average genome coverage was ∼96%. The 17 chromosomes are portrayed along the perimeter of the outer circle. Insertions (purple), deletions (blue), deletions which contain an insertion (orange) or inversions (red) are represented in the second circle. The third circle represents the distribution of SNP density (green), i.e., the number of SNPs within 100 kb of each dot. The innermost circle indicates intrachromosomal and interchromosomal (gray) transfer, indicated by connections between segments.
‘Indo’ exhibited greater sequence variation than ‘Su Shuai’ with respect to the ‘Gold Delicious’ reference genome. The genome-wide mutation rate for ‘Indo’ was 1 change per 220 bases versus 1 change per 298 bases in ‘Su Shuai’. Among the 17 chromosomes (chr) that comprise the apple genome, the highest number of SNPs was observed in chr 2 in both ‘Indo’ and ‘Su Shuai’, with one change per 186 bases on chr 2 of ‘Indo’ and one change per 235 bases on chr 2 of ‘Su Shuai’. Notably, chr 7 in both ‘Indo’ and ‘Shu Shuai’ exhibited the lowest level of mutations, with one SNP per 378 bases and one SNP per 278 bases, respectively (Supplementary Files 1 and 2).
SnpEff 3.0f was used to further evaluate the SNPs that Samtools had detected based on the annotation of the apple reference genome. Approximately 93.58% of the identified SNPs were considered sequence modifiers; the remaining (∼6.42%) consisted of high impact (∼0.15%), moderate impact (∼3.05% avg.) or low impact (∼3.22% avg.) changes in the transcriptional unit. In the current study, a total of 6578 SNPs were considered high impact SNPs in ‘Indo’ (0.144% of the total number of SNPs) and 5193 in ‘Su Shuai’ (0.155%). Further examining the three possible effects in terms of functional classes (missense, nonsense, and silent) in ‘Indo’ and ‘Su Shuai’ identified 51.47% and 50.42% missense SNPs, 47.15% and 48.27% silent SNPs, and a small proportion of nonsense SNPs, 1.384% and 1.303%, respectively. The missense/silent ratio for ‘Indo’ was 1.092% and 1.0445% for ‘Su Shuai’ (Supplementary Files 1 and 2).
SVs
In addition to SNPs, the prevalence of InDels and some other types of larger SVs was assessed. SV data sets were obtained by integrating the results from Pindel, BreakDancer and several other software programs. Approximately 95% of the SVs identified in the two cultivars were insertion (INS) and deletion (DEL) variants, and the remaining 5% consisted of deletions that contained an inverted insertion (INV), an intrachromosome transfer (ITX) an interchromosome transfer (CTX) and deletions which contain insertion (IDE). The number of SVs in ‘Indo’ was 59 547, of which InDels accounted for 95.08% of the total, 0.71% were IDE, 0.06% were INV, 0.39% were ITX and 3.76% were CTX. A total of 50 143 SVs were identified in ‘Su Shuai’, of which InDels accounted for 94.56% of the total, 0.64% were IDE, 0.08% were INV, 0.42% were ITX and 4.30% were CTX (Table 5).
The majority of variants were found within intergenic and repeat regions. The distributions of SVs in different components of the genome were determined (Table 4). In ‘Indo’, the number of SVs in exons was approximately 3261 (comprising 5.5% of the total), 14 758 (24.8%) occurred in introns and 30 134 (50.6%) were located in intergenic regions. The distribution of SVs in ‘Su Shuai’ was similar to ‘Indo’, in which 2741 (5.5%) were located in exons, 12 174 (24.3%) in introns and 25 127 (50.1%) in intergenic regions (Table 4). Some InDels were predicted to be frameshift mutations, but the lengths of InDels in coding regions are more likely to be multiples of 3 (the length of a codon) compared with InDels in the rest of the genome (Figure 3).

A comparison of the distributions of InDels in the whole genome and the coding (CDS) region of the genome. (a) ‘Indo’; (b) ‘Su Shuai’. The numbers of Insertions and Deletions in the genome are shown in yellow and red, respectively. The numbers of Insertions and Deletions within the CDS are shown in blue and green, respectively. The x-axis represents InDel size. The left scale represents the number of frameshift mutations in the genome and the right scale represents the number of frameshift mutations in the CDS.
Functional annotation of gene sequences obtained for ‘Indo’ and ‘Su Shuai’
Three kinds of gene transcripts can result from the presence of a SNP or SV. Non-synonymous (NS) mutations caused by SNPs lead to a change in the sequence of the translated protein, frameshifts (F) resulting from InDel mutations in genes lead to a change or loss of gene function, and SV modifications that occur in a gene result in changes in the structure and function of its encoded protein. NS mutations, F mutations and SVs can all cause changes in gene function.
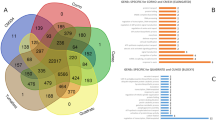
In total, 24 449 and 22 153 modified genes were identified in ‘Indo’ and ‘Su Shuai’, respectively, based on alignment of their DNA sequences to the ‘Golden Delicious’ reference genome (Figure 4a). Further analysis indicated that 825 of the modified genes were ‘Su Shuai’-specific and 3121 of the modified genes were ‘Indo’-specific (Figure 4b). In the genome sequence data obtained from ‘Indo’, a total of 22 424 genes with NS mutations were identified, of which 5121 were genes with (non-synonymous loci ≥10) NS, 1407 were genes with an F arising from an InDel, and 2882 were genes with a SV. In ‘Su Shuai’, a total of 19 953 genes with NS mutations were identified, of which 4097 were NS (≥10), 1216 were genes with an F and 2419 were genes with a SV (Table 6).

Flowchart for finding genes potentially associated with horticultural traits in ‘Su Shuai’. (a) Venn diagram of unigenes in ‘Golden Delicious’ (G), ‘Indo’ (I) and ‘Su Shuai’ (S). (b) Venn diagram illustrating the overlap of unigenes obtained from ‘Su Shuai’ (S) and ‘Indo’ (I). A total of 24 449 unigenes were obtained from ‘Indo’ and 22 153 from ‘Su Shuai’. A total of 3121 genes were ‘Indo’-specific and 825 were specific to ‘Su Shuai’. (c) After screening to identify gene variants associated with horticultural traits in ‘Su Shuai’, 17 genes related to disease resistance, 10 genes related to gibberellin (GA) and 19 genes related to sorbitol metabolism were found in both ‘Indo’ and ‘Su Shuai’, including only non-synonymous mutations.
Approximately 73% of the differential genes (17 726 for ‘Indo’ and 16 201 for ‘Su Shuai’) were classified by Pfam using an E-value ≤1e−5. 2803 gene families in ‘Indo’ and 2748 in ‘Su Shuai’ were classified by Pfam. Many gene families were categorized as Pkinase domains (Family) for both ‘Indo’ and ‘Su Shuai’. LRRNT_2 (256 genes in ‘Indo’ and 241 genes in ‘Su Shuai’) and PPR Family (256 genes in ‘Indo’ and 232 genes in ‘Su Shuai’) were the second most abundant categories. In this study, significant gene families were identified using a threshold e-value of 10−5 (Supplementary Files 3 and 4).
A total of 5216 and 4736 unigenes from ‘Indo’ and ‘Su Shuai’, respectively, mapped to 118 different KEGG pathways. The pathways with the highest representation in ‘Indo’ were ubiquinone and other terpenoid–quinone biosynthesis pathways (ko00130, 223 unigenes, 4.0%), followed by the phosphate pathway (ko00030, 179 unigenes, 3.2%) and folate biosynthesis (ko00790, 173 unigenes, 2.1%). The highest represented pathways in ‘Su Shuai’ were pyruvate metabolism (ko00620, 198 unigenes, 4.0%), followed by pyrimidine metabolism (ko00240, 162 unigenes, 3.2%) and nitrogen metabolism (ko00910, 159 unigenes, 3.2%) (Supplementary Files 5 and 6).
In the current study, three horticultural traits (disease resistance, internode length and fruit flavor) were evaluated. A total of 260 disease resistance-related genes were identified in ‘Su Shuai’, including 218 genes with a NS mutation, 19 genes with an S/NS/SV (NS mutations in SNPs and SVs) mutation, and 10 genes with an S/NS/SV/F mutation. We also identified 716 (TIR)-NBS-LRR genes, of which 591 had NS mutations, 48 had S/NS/SV mutations, 42 had S/NS/SV/F mutations, 2 had a SV mutation and 2 had a SV/F mutation.
Regarding internode length, 429 gibberellin-related genes were identified, of which 353 had a NS mutation, 13 had an S/NS/SV mutation and 18 had an S/NS/SV/F mutation. Five of the gibberellin-related genes had a SV mutation and one had an SV/F mutation.
Seventeen disease resistance and LRR genes in ‘Su Shuai’ displayed differences in their levels of expression compared with their expression in ‘Golden Delicious’ and ‘Indo’. Five mRNAs were associated with gene IDs encoding for products categorized as disease resistance proteins (N, TIR-NBS-LRR type R protein 7). Twelve mRNAs encoded products related to LRR receptor-like serine/threonine-protein kinase or LRR protein (Table 7). Two GA-related genes, GA3OX1 and RGL2, and eight GA synthesis and GA signal regulation genes, were found to be differentially expressed (Table 8). Nineteen genes associated with carbohydrate metabolism were identified as being differentially expressed. One of these genes related to NAD-dependent malic enzyme 2 had a S/NS/SV mutation, and one gene related to alkaline/neutral invertase CINV2 also had a S/NS/SV mutation. Seven mRNAs, encoding sorbitol dehydrogenase (SDH), all had NS mutations. Eight sorbitol transporter genes were differentially expressed, among which one had an S/NS/SV/F mutation and seven had a NS mutation. One gene (S6PDH, MDP0000408705) encoding NADP-dependent D-sorbitol-6-phosphate dehydrogenase and one gene (MDP0000312001) involved in NADPH-dependent aldose-6-phosphate reductase activity (GO: 0047641, annotation from Gene Ontology database) had NS mutations (Table 9 and Figure 4c).
RT-qPCR analysis to assess differential gene expression
To assess whether the genes were differentially expressed, eight genes were selected for RT-qPCR analysis. The expression levels of each gene in ‘Golden Delicious’, ‘Indo’ and ‘Su Shuai’ were compared. The relative expression levels of the genes were calculated using the 2−ΔCTΔCT method (Figure 5). The genes associated with disease resistance selected for the analysis included the putative disease resistance family protein gene N (MDP0000272916) and the TIR-NBS-LRR type R protein 7 MbR7 (MDP0000233155). Genes related to gibberellin metabolism included GA3OX1 (MDP0000233590), RGL2 (MDP0000300311) encoding gibberellin 3-beta-dioxygenase 1, the DELLA protein RGL2, and a gene (MDP0000284679) related to the negative regulation of the GA-mediated signaling pathway. Genes associated with carbohydrate metabolism included the sorbitol dehydrogenase gene SDH (MDP0000693768), CINV2 (MDP0000745777) encoding alkaline/neutral invertase CINV2 and a homolog of NAD-ME2 (MDP0000453139) encoding a NAD-dependent malic enzyme 2.

RT-qPCR of analysis of gene expression in ‘Su Shuai’ and ‘Indo’, relative to ‘Golden Delicious’. Specific genes were selected based on their associations with different horticultural traits (disease resistance, internode length and fruit flavor). All RT-qPCR analyses were repeated three times (biological replicates). Apple Tubulin, Mdtubulin was used as a housekeeping gene (GenBank accession number AJ421411). The x-axis indicates the different genotypes. The y-axis shows the relative expression level as determined by RT-qPCR. The level of gene expression in ‘Golden Delicious’ was arbitrarily assigned a value of 1.0.
Results of the RT-qPCR analysis indicated that the expression of N and MbR7 was highest in ‘Su Shuai’. The expression of GA3OX1 and RGL2 was highest in ‘Golden Delicious’. The expression of the gene (MDP0000284679) associated with the negative regulation of GA-mediated signaling, however, was the highest in ‘Su Shuai’, followed by ‘Indo’ and the lowest in ‘Golden Delicious’. Regarding the genes associated with carbohydrate metabolism, the expression of SDH was the highest in ‘Su Shuai’, followed by ‘Indo’ and the lowest in ‘Golden Delicious’. NAD-ME2 had a higher level of expression in ‘Indo’ and lower levels of expression in ‘Golden Delicious’ and ‘Su Shuai’. The expression of CINV2 was the highest in ‘Golden Delicious’ and low in ‘Indo’ and ‘Su Shuai’. Results of the RT-qPCR analysis were in general concordant with the phenotypic differences that were observed among the three cultivars.
Discussion
Minor variants and structural variants represent different types of genomic modifications. Although natural selection acts on both types of variants, crop breeding efforts primarily target minor variants such as SNPs, because their inheritance patterns are better understood and, therefore, more efficiently manipulated, and because minor variants code for single potential changes in amino acids that may or may not result in a functional change in the coded protein. An improved understanding of the processes by which structural variants occur, their locations and their phenotypic effects is now possible through advanced genomic methods.6
SNP variants are more abundant than SV variants in ‘Indo’ and ‘Su Shuai’
The SNP ratios (SNP/bp) observed in this study were typically between 1/100 and 1/300 bp. This observation is consistent with the average of 4.4 SNPs per 1000 bp reported for the apple genome and previous results observed in other crop plants.3,32 The cultivar ‘Indo’ had a larger SNP/bp ratio (1/220) compared with ‘Su Shuai’ (1/298). The SNP variant event ratio was lower for ‘Su Shuai’ than for ‘Indo’ because ‘Golden Delicious’ was used as the male parent of ‘Su Shuai’ (Figure 1).
‘Indo’ also exhibited a lower proportion of heterozygous versus homozygous variants (79.09%) compared to ‘Su Shuai’ (89.82%). This situation may be a consequence of changes in gene sequences that occurred during hybridization. Variation in the rates of change between chromosomes (chrs) and within chrs is, in part, influenced by the length of the chr.33 Chr 2, however, exhibited the highest rate of sequence modifications in both genotypes, despite not being the largest chr. The ranking of chrs from the shortest to the longest based on the sequence of the ‘Golden Delicious’ apple reference genome sequence is: chr 16, chr 4, chr 17, chr 6, chr 7, chr 14, chr 8, chr 1, chr 12, chr 9, chr 5, chr 10, chr 13, chr 3, chr 11, chr 2 and chr 15.
The high rate of variation observed in chr 2 may be because of the presence of a greater number of recombination hotspots, as has been previously reported by Nachman for humans.34 Chr 2 has been reported to carry important QTL for the nucleotide binding site (NBS) encoding domain of plant disease resistance genes and includes 13 NBS markers.35 Chr 2 contains more NBS and LRR (leucine-rich repeats)-kinase genes (∼420) than any of the other apple chromosomes. Disease resistance is an important target for selection. Ulrich and Dunemann identified a candidate gene within a volatile compound QTL mapped to chr 2.36,37 Notably, ‘Su Shuai’s’ fruit has little aroma. This observation suggests that some of the genes with SNP variants that were identified in the current study may be involved in the synthesis and metabolism of volatile substances. A high level of recombination does not necessarily represent a source of new, functional alleles, because recombination hotspots often occur within intergenic regions in plants32,38 and their distribution along the chromosome is influenced by several factors, including proximity to the centromere, gene density and GC content.39 A better understanding of the distribution of these hotspots will lead to better modeling of the inheritance and conformations of linkage blocks.
Even silent changes within an exon are capable of affecting the structure and function of the resultant protein.40 Regarding the proportion of silent changes (approximately 48%) and missense modifications (approximately 51%) in the two genotypes, ‘Indo’ exhibited a value of 1.0917 for missense/silent, which was similar to the value of 1.0449 observed in ‘Su Shuai’. The transition/transversion (Ts/Tv) was approximately 2.1 for the two genotypes. Ts/Tv ratios have been variously estimated to be 3.9, 3.6, 1.9, 1.6 and 2.5 for other plants, such as maize, alfalfa (Medicago sativa L.), einkorn wheat (Triticum monococcum L.), barley (Hordeum vulgare L.) and plants in the genus Lotus, respectively.41 Information regarding Ts/Tv in species related to apple and other crops in general is scarce. Ts/Tv is a theoretical estimator of mutation rates and divergence, but is not directly related to observed rates of change at the phenotypic level.42 Our collective analyses indicate that ‘Su Shuai’ presents greater similarity to its female parent ‘Indo’ in terms of gene loci (all genes and candidate genes with horticultural value).
‘Indo’ and ‘Su Shuai’ exhibited the same types of the most common amino acid substitutions: threonine to alanine, valine to alanine and isoleucine to valine. Codon substitution, amino-acid substitution and protein features have been routinely used to describe patterns of differentiation among organisms as a way of reconstructing phylogenetic relationships.43 Amino-acid substitution (also known as amino acid replacement) is often the preferred parameter used in comparative genomics, because it provides functional information regarding the effects of the substitutions and does not suffer from codon bias or problems associated with post-transcriptional modification.6
Nucleotide and amino acid substitutions have been shown to affect important horticultural traits. Barry et al.44 identified two specific amino-acid substitutions that affect the degradation of green color in tomato. The effects of these amino acid substitutions on the characteristics of the protein suggest that their substitutions may be responsible for the relevant biological consequences. Vogt et al.45,46 reported that the AvrRpt2EA type III effector plays a significant role in the activation of a potential resistance pathway in Malus robusta 5. Importantly, a SNP resulting in an amino acid change from cysteine (C) to serine (S) at position 156 in the AvrRpt2EA protein sequence was correlated with the severity of virulence of Erwinia amylovora strains, the causal agent of fire blight, to Malus × robusta 5.45,46
In the present study, approximately 95% of SVs reported in the two genotypes were InDels, and the remaining 5% were IDE, INV, ITX and CTX. Therefore, the focus of our analysis was on small and large InDels, rather than IDE, INV, ITX and CTX variants. In ‘Su Shuai’, deletions represented the majority of InDels present, whereas in ‘Indo’, a similar number of insertions and deletions were present. InDels can be scattered throughout a genome and can affect exon regions, as has been evidenced in humans,47,48 cattle,49 silkworm50 and Arabidopsis.51 We identified thousands of small InDels, some of which cause protein-coding frameshifts and others that occur in parts of genes that may result in modifications to gene regulatory functions. By contrast to SNPs identified in our study, relatively few InDels are located within the coding regions of genes, and when they do occur within an exon, the majority are trinucleotide InDels. These observations mirror findings in other species49,52 and are not surprising if there is selection against InDels that disrupt protein sequences, such as frameshift mutations within exons. Confirming the expected location of InDels also provides confidence in the set of InDels that we identified. We cannot exclude, however, that some frameshift mutations are because of incorrect gene prediction models or that some InDels are sequencing errors. Assuming that these possibilities have minimal effect on the overall number of identified InDels, our data demonstrate the major contribution that SNPs contribute to genetic variation in ‘Su Shuai’, but that InDels may also play an important role in the modification of protein-coding regions.
Homologous genes linked to disease resistance
In plants, the LRR domain conveys recognition specificity to an infecting pathogen and binds to a corresponding ligand with a putative NB site. Genes with these motifs are termed ‘NB-LRR’ genes.53 ‘NB-LRR’ genes include members that carry either N-terminal homology to the Toll protein and interleukin-1 receptor (TIR-NB-LRR) or a putative coiled-coil at the N-terminus (CC-NB-LRR). Genes from both of these subclasses confer resistance against fungi. As a result, these genes have been labeled as resistance (R) genes and several R-genes have been used in crop improvement programs. NB-LRR genes that confer resistance against flax rust, maize rust, barley powdery mildew, rice blast and Fusarium wilt, and downy mildew of tomato have been identified.54,55 Plant NB-LRRs, directly or indirectly, recognize specific pathogenic virulence factors or effectors and trigger a robust defense response which often manifests in localized cell death, known as a hypersensitive response. The truncated or single mutation derivatives of plant NB-LRRs could have an important influence on defense response to plant pathogens.56 Among plant genomes, apple has the highest number of predicted genes in its genome (57 386, including some genes that may be present in only one of the two chromosomes in a pair) and the total number of NBS resistance genes identified in apple is 992, of which 58% are NBS-LRR and 27% are TIR-NBS-LRR genes.17
Based on the above results, five disease resistance genes were identified in ‘Su Shuai’, four of which are homologous to the N encoding for tobacco mosaic virus (TMV) resistance protein. All four of these genes contain S/NS/SV variation in their sequence with respect to ‘Golden Delicious’, whereas S/NS variation was present in ‘Indo’ (Table 7). One of the disease resistance genes is homologous to the MbR7 gene, which encodes a TIR-NBS-LRR type R protein 7 that was annotated in the NR protein database. The MbR7 gene has an S/NS/S/F mutation in ‘Su Shuai’, with respect to ‘Golden Delicious’, whereas ‘Indo’ exhibits a S/NS variation (Table 7). Four of the identified R-genes are homologous to the N gene that encodes TMV resistance protein, which triggers a defense system and a hypersensitive response.52,57 The dominant TMV resistance gene N is a member of the TIR-NB-LRR class of resistance genes.58 The N gene encodes two alternatively spliced mRNAs that possess a TIR domain, a NB domain, and a LRR, all of which are required for N function.59,60 RT-qPCR analysis indicated that the N gene had a higher level of expression in ‘Su Shuai’ compared with ‘Golden Delicious’ and ‘Indo’. The MbR7 homologous gene (MDP0000233155) encodes a TIR-NBS-LRR type R protein 7. Lee et al.61,62 reported that the ectopic expression of an apple TIR-NBS-LRR R gene MbR7 in heterologous transgenic Arabidopsis enhanced resistance to Pseudomonas syringae pv. tomato DC3000 infection. Ma et al.63 reported that the expression of Md-NBS was significantly higher in varieties resistant to apple leaf spot disease, suggesting that Md-NBS may play an important role in resistance to this pathogen. Similarly, our results indicated that the expression of MbR7 was significantly higher in ‘Su Shuai’ than in ‘Golden Delicious’ and ‘Indo’, both of which are susceptible to Alternaria leaf spot. These data suggest that the homologous MbR7 gene may play an important role in resistance to apple leaf spot disease. Mutations in the sequence of the MbR7 homologous gene may affect the level of disease resistance conferred by this gene.
The remainder of the LRR-domain genes identified in the present study encoded LRR receptor-like serine/threonine-protein kinases or LRR proteins, which also have been associated with pathogen defense, transduction and regulation of signaling, and control of cell population size in the shoot apical meristem.64 Sequence variation within the central LRR domain and variation in LRR copy number play important roles in determining recognition specificity.65 Likewise, R genes, which were first identified as dominant resistance genes, could be targets of pathogen effectors.66 Therefore, if their sequences are modified, resistance of plants to pathogens may be altered. These sequence modifications may manifest as a loss in disease-resistance functions for some genes or alternatively as increases in resistance functions. The specific effects of sequence variations in these genes on their functions will require further research.
GA-associated genes are potentially involved in regulating internode length
GA plays an important role in plant growth and development, particularly in determining internode length.67 DELLA proteins, which were named for the conserved order of aspartic acid (D), glutamic acid (E), leucine (L) and alanine (A) at the N-terminus, are negative regulators of the GA signaling pathway.68
Ten GA synthesis, transport and signal transduction-related genes were identified in ‘Su Shuai’ with striking variations in their sequences relative to their sequences in ‘Golden Delicious’ and ‘Indo’. Two of the genes were annotated as homologous to the GA3OX1 gene (MDP0000233590, with S/NS/Sv mutations), which codes for Gibberellin 3-beta-dioxygenase 1, and the RGL2 gene (MDP0000300311, with S/Sv/F mutations), which codes for the DELLA protein RGL2. Gibberellin 3-beta-dioxygenase 1 is involved in converting the inactive GA precursors, GA9 and GA20, into the bioactive gibberellins, GA4 and GA1, which are involved in vegetative growth and development. The double mutant ga3ox1/ga3ox2 exhibits a severe defect in seed germination and root growth and has a dwarf phenotype.69–71 The DELLA protein RGL2 is most likely a transcriptional regulator that acts as a repressor of the GA signaling pathway. RGL2 most likely acts as a member of several large protein complexes that repress the transcription of GA-inducible genes. Upon exogeneous application of GA, RGL2 is degraded within the proteasome, allowing the GA signaling pathway to function. The activities of RGL2 and other DELLA proteins are most likely regulated by other phytohormones such as auxin and ethylene.72–74 In the present study, RT-qPCR analysis indicated that MDP0000233590 (homologous to GA3OX1) and MDP0000300311 (homologous to RGL2) both had low expression in ‘Su Shuai’ relative to ‘Golden Delicious’ and ‘Indo’.
Of the remaining eight genes annotated in biological process by the GO database, one (MDP0000284679, with an S/SV/F mutation) is associated with the negative regulation of the GA-mediated signaling pathway (GO: 0009938), three are associated with the GA biosynthetic process (GO: 0009686) and four are associated with response to GA stimulus (GO: 0009739). All of these eight genes had higher levels of expression in ‘Su Shuai’ relative to ‘Golden Delicious’ and ‘Indo’. We infer that the phenotype of short internodes seen in ‘Su Shuai’ may be because of the mutations present in these genes.
The dwarf phenotype in ‘Su Shuai’, however, is not necessarily only because of the mutations present in the above genes. Although these genes have expression differences between ‘Su Shuai’ and its parents (‘Golden Delicious’ and ‘Indo’), dwarfism can also be caused by blockage in the GA signaling pathway or by defects in other plant hormones that regulate plant form, such as brassinosteroids or strigolactone.68,75 Foster et al.76 found that the largest class of genes significantly upregulated in dwarfing rootstocks were associated with response to biotic and abiotic stress, suggesting that stress, possibly mediated by JA and ABA signaling, also plays a role in the dwarfing phenotype.
Carbohydrate metabolism-related candidate genes are possibly involved in fruit flavor
Carbohydrate metabolism plays an important role in defining fruit composition and the accumulation of sugars has a great effect on fruit flavor. Compared with other plant genomes, apple has considerably more copies of key genes related to sorbitol metabolism. These genes include aldose 6-P reductase, which is rate-limiting for sorbitol biosynthesis, and sorbitol-dehydrogenase (SDH), which converts sorbitol to fructose in the fruit.77 Soluble sugars and organic acids are important components of fruit taste and, combined with aroma, define the quality of apples. Fruit taste depends on the types and levels of soluble sugars and organic acids. In mature apples, as in most fleshy fruits, the main soluble sugars are fructose, glucose and sucrose. The main organic acids are malic and citric acids.78 In Rosaceae, photosynthesis-derived carbohydrates are transported mainly as sorbitol.79 We identified one gene (MDP0000693768) homologous to SDH, which encodes sorbitol dehydrogenase. The sequence of this gene had four NS point mutations in ‘Su Shuai’, whereas no mutations were observed in ‘Indo’. Notably, the expression of genes homologous to SDH was lower in ‘Su Shuai’ relative to ‘Golden Delicious’ and ‘Indo’. In the present study, we did not identify genes related to sorbitol metabolism with SV mutations. One homolog to CINV2 (MDP0000745777) was detected that encodes the alkaline/neutral invertase CINV2, which may cleave sucrose into glucose and fructose.80 One SV mutation was also found in a gene (MDP0000453139) homologous to NAD-ME2, encoding NAD-dependent malic enzyme 2, which might be involved in the regulation of sugar and amino acid metabolism during the night period. When associated with NAD-ME2 disruption, loss of NAD-dependent malic enzyme activity is associated with an altered steady-state level of sugars and amino acids at the end of the light period.81 The expression levels of genes homologous to SDH were low in ‘Su Shuai’ and ‘Indo’ relative to ‘Golden Delicious’.
Conclusions
Based on the genome sequence data obtained by re-sequencing ‘Su Shuai’ and ‘Indo’, we identified SNPs, SVs and genes associated with trait characteristics. Substantial variation was detected in the genome sequences of both ‘Su Shuai’ and ‘Indo’ despite sampling only two individuals. Many unigenes were identified and annotated, some of which appeared to be cultivar specific. These data provide an excellent platform for future genetic and functional genomic research in these cultivars and in apple in general. Genes related to disease resistance, short internodes and lighter fruit flavor, and their expression levels in ‘Golden Delicious’, ‘Indo’ and ‘Su Shuai’ were analyzed by RT-qPCR. These analyses provided new insights into the molecular mechanisms underlying several ‘Su Shuai’ horticultural traits. Variation in the gene sequence for MbR7 (MDP0000233155) encoding a TIR-NBS-LRR type R protein 7 may play a role in disease resistance in ‘Su Shuai’, particularly in resistance to the Alternaria leaf spot. The DELLA protein RGL2 homolog (MDP0000300311) and a gene related to the negative regulation of the GA-mediated signaling pathway (MDP0000284679) may be involved in the short internodes observed in ‘Su Shuai’ based on their differences observed in ‘Su Shuai’ relative to ‘Golden Delicious’ and ‘Indo’. The gene homologous to SDH associated with sorbitol metabolism (MDP0000693768), and the gene homologous to NAD-ME2 associated with accumulation and loss of organic acids during fruit ripening (MDP0000453139) may be involved in fruit flavor in ‘Su Shuai’. Collectively, these data provide important information on genetic variation in ‘Su Shuai’ that may facilitate the molecular-assisted breeding of apples.
References
Edwards D, Batley J . Plant genome sequencing: applications for crop improvement. Plant Biotechnol J 2010; 8: 2–9.
Van K, Rastogi K, Kim K, Lee S . Next-generation sequencing technology for crop improvement. Sabrao J Breed Genet 2013; 45: 84–99.
Consortium EP . An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489: 57–74.
Imelfort M, Edwards D . De novo sequencing of plant genomes using second-generation technologies. Brief Bioinform 2009; 10: 609–618.
Alkan C, Coe BP, Eichler EE . Genome structural variation discovery and genotyping. Nat Rev Genet 2011; 12: 363–376.
Fresnedo-Ramírez J, Martínez-García PJ, Parfitt DE, Crisosto CH, Gradziel TM . Heterogeneity in the entire genome for three genotypes of peach [Prunus persica (L.) Batsch] as distinguished from sequence analysis of genomic variants. BMC Genomics 2013; 14: 750–716.
Di Genova A, Almeida AM, Mun C et al. Whole genome comparison between table and wine grapes reveals a comprehensive catalog of structural variants. BMC Plant Biol 2014; 14: 7–19.
Beckmann JS, Estivill X, Antonarakis SE . Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet 2007; 8: 639–646.
Goettel W, Messing J . Divergence of gene regulation through chromosomal rearrangements. BMC Genomics 2010; 11: 678–697.
Springer NM, Ying K, Fu Y et al. Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content. PLoS Genet 2009; 5: e1000734.
Swanson-Wagner RA, Eichten SR, Kumari S et al. Pervasive gene content variation and copy number variation in maize and its undomesticated progenitor. Genome Res 2010; 20: 1689–1699.
DeBolt S . Copy number variation shapes genome diversity in Arabidopsis over immediate family generational scales. Genome Biol Evol 2010; 2: 441–453.
Haun WJ, Hyten DL, Xu WW et al. The composition and origins of genomic variation among individuals of the soybean reference cultivar ‘Williams 82’. Plant Physiol 2011; 155: 645–655.
McHale LK, Haun WJ, Xu WW et al. Structural variants in the soybean genome localize to clusters of biotic stress-response genes. Plant Physiol 2012; 159: 1295–1308.
Peace C, Norelli J . Genomics approaches to crop improvement in the Rosaceae. In: Peace C, Norelli J . (ed.) Genetics and genomics of Rosaceae. 1st ed. New York: Springer, 2009: 19–53.
Hummer KE, Janick J . Rosaceae: taxonomy, economic importance, genomics. In: Folta K M, Gardiner SE (ed.) Genetics and Genomics of Rosaceae. 2nd ed. New York: Springer, 2009: 1–17.
Velasco R, Zharkikh A, Affourtit J et al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat Genet 2010; 42: 833–839.
Troggio M, Gleave A, Salvi S et al. Apple, from genome to breeding. Tree Genet Genomes 2012; 8 (3): 509–529.
Sheng BC, Qu SC . [A new apple cultivar ‘Su Shuai’.] Acta Horticul 2012; 39: 2075–2076. Chinese.
Liu Z, Wang DM, Zhang JE, Yi K, Yang F, Yan ZY . [A new middle-late ripening apple cultivar ‘Yueyanghong’.] Acta Hort 2009; 36: 1545–1546. Chinese.
Yan ZL, Zhang HT, Guo GN, Zhang SN, Liu ZZ . [A new middle-late ripening apple cultivar ‘Jinxiu Hong’.] Acta Hort 2010; 9: 1531–1532. Chinese.
Tong ZG, Wang FR, Zhang Z et al. [A method for DNA extraction from mature leaves of fruit trees.] J Fruit Sci 2008; 25: 122–125. Chinese.
Li H, Handsaker B, Wysoker A et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
Li H, Durbin R . Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010; 26: 589–595.
Li H, Durbin R . Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
Ahmad R, Parfitt DE, Fass J et al. Whole genome sequencing of peach (Prunus persica L.) for SNP identification and selection. BMC Genomics 2011; 12: 569–576.
Cingolani P, Platts A, Wang LL et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain ‘w1118’; ‘iso-2’; ‘iso-3’. Fly (Austin) 2012; 6: 80–92.
Ye K, Schulz MH, Long Q, Apweiler R, Ning Z . Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009; 25: 2865–2871.
Chen K, Wallis JW, McLellan MD et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 2009; 6: 677–681.
Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M . The KEGG resource for deciphering the genome. Nucleic Acids Res 2004; 32: D277–D280.
Finn RD, Bateman A, Clements J et al. Pfam: the protein families database. Nucleic Acids Res 2013; gkt1223: 1–9.
Mezard C . Meiotic recombination hotspots in plants. Biochem Soc Trans 2006; 34: 531–534.
Fledel-Alon A, Wilson DJ, Broman K et al. Broad-scale recombination patterns underlying proper disjunction in humans. PLoS Genet 2009; 5: e1000658.
Nachman MW . Single nucleotide polymorphisms and recombination rate in humans. Trends Genet 2001; 17: 481–485.
Calenge F, van der Linden C, van de Weg E et al. Resistance gene analogues identified through the NBS-profiling method map close to major genes and QTL for disease resistance in apple. Theor Appl Genet 2005; 110: 660–668.
Ulrich D, Dunemann F . Towards the development of molecular markers for apple volatiles. Flavour Fragr J 2012; 27: 286–289.
Potts SM, Khan MA, Han Y, Kushad MM, Korban SS . Identification of quantitative trait Loci (QTLs) for fruit quality traits in apple. Plant Mol Biol Report 2014; 32: 109–116.
Schnable PS, Hsia AP, Nikolau BJ . Genetic recombination in plants. Curr Opin Plant Biol 1998; 1: 123–129.
Paape T, Zhou P, Branca A, Briskine R, Young N, Tiffin P . Fine-scale population recombination rates, hotspots, and correlates of recombination in the Medicago truncatula genome. Genome Biol Evol 2012; 4: 726–737.
Komar AA . SNPs, silent but not invisible. DNA 2007; 315: 466–467.
Vitte C, Bennetzen JL . Analysis of retrotransposon structural diversity uncovers properties and propensities in angiosperm genome evolution. Proc Natl Acad Sci USA 2006; 103: 17638–17643.
Yang Z, Yoder AD . Estimation of the transition/transversion rate bias and species sampling. J Mol Evol 1999; 48: 274–283.
Lio P, Goldman N . Models of molecular evolution and phylogeny. Genome Res 1998; 8: 1233–1244.
Barry CS, McQuinn RP, Chung MY, Besuden A, Giovannoni JJ . Amino acid substitutions in homologs of the STAY-GREEN protein are responsible for the green-flesh and chlorophyll retainer mutations of tomato and pepper. Plant Physiol 2008; 147: 179–187.
Vogt I, Wöhner T, Richter K et al. Gene-for-gene relationship in the host-pathogen system Malus × robusta 5-Erwinia amylovora. New Phytol 2013; 197: 1262–1275.
Broggini GA, Wöhner T, Fahrentrapp J et al. Engineering fire blight resistance into the apple cultivar ‘Gala’ using the FB_MR5 CC-NBS-LRR resistance gene of Malus × robusta 5. Plant Biotechnol J 2014; 12: 728–733.
Mullaney JM, Mills RE, Pittard WS, Devine SE . Small insertions and deletions (INDELs) in human genomes. Hum Mol Genet 2010; 19: R131–R136.
Mills RE, Pittard WS, Mullaney JM et al. Natural genetic variation caused by small insertions and deletions in the human genome. Genome Res 2011; 21: 830–839.
Zhan B, Fadista J, Thomsen B, Hedegaard J, Panitz F, Bendixen C . Global assessment of genomic variation in cattle by genome resequencing and high-throughput genotyping. BMC Genomics 2011; 12: 557.
Xia Q, Guo Y, Zhang Z et al. Complete resequencing of 40 genomes reveals domestication events and genes in silkworm (Bombyx). Science 2009; 326: 433–436.
Cao J, Schneeberger K, Ossowski S et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet 2011; 43: 956–963.
Dinesh-Kumar S, Baker BJ . Alternatively spliced N resistance gene transcripts: their possible role in tobacco mosaic virus resistance. Proc Natl Acad Sci USA 2000; 97: 1908–1913.
Cuenca J, Aleza P, Vicent A, Brunel D, Ollitrault P, Navarro L . Genetically based location from triploid populations and Gene Ontology of a 3.3-Mb genome region linked to Alternaria brown spot resistance in citrus reveal clusters of resistance genes. PLoS ONE 2013; 8: e76755.
Dangl JL, Jones JD . Plant pathogens and integrated defence responses to infection. Nature 2001; 411: 826–833.
Perazzolli M, Malacarne G, Baldo A et al. Characterization of resistance gene analogues (RGAs) in apple (Malus × domestica Borkh.) and their evolutionary history of the rosaceae family. PLoS ONE 2014; 9: e83844.
Mukhtar MS . Engineering NLR immune receptors for broad-spectrum disease resistance. Trends Plant Sci 2013; 18: 469–472.
Dinesh-Kumar S, Tham WH, Baker BJ . Structure–function analysis of the tobacco mosaic virus resistance gene N. Proc Natl Acad Sci USA 2000; 97: 14789–14794.
Bent AF, Mackey D . Elicitors, effectors, and R genes: the new paradigm and a lifetime supply of questions. Annu Rev Phytopathol 2007; 45: 399–436.
Caplan J, Padmanabhan M, Dinesh-Kumar SP . Plant NB-LRR immune receptors: from recognition to transcriptional reprogramming. Cell Host Microbe 2008; 3: 126–135.
Niemeyer J, Ruhe J, Machens F, Hehl R . Differential expression of the TMV resistance gene N prevents a hypersensitive response in seeds and during germination. Planta 2013; 237: 909–915.
Lee SY, Choi YJ, Ha YM, Lee DH . Ectopic expression of apple MbR7 gene induced enhanced resistance to transgenic Arabidopsis plant against a virulent pathogen. J Microbiol Biotechnol 2007; 17: 130–137.
Zheng HQ, Zhang Q, Li HX, Lin SZ, An XM, Zhang ZY . Over-expression of the triploid white poplar PtDrl01 gene in tobacco enhances resistance to tobacco mosaic virus. Plant Biology 2011; 13: 145–153.
Ma C, Lu Y, Bai S et al. Cloning and characterization of miRNAs and their targets, including a novel miRNA-targeted NBS–LRR protein class gene in apple (Golden Delicious). Mol Plant 2014; 7: 218–230.
Wang GD, Long YC, Thomma BP, de Wit PJ, Angenent GC, Fiers M . Functional analyses of the CLAVATA2-like proteins and their domains that contribute to CLAVATA2 specificity. Plant Physiol 2010; 152: 320–331.
Gururani MA, Venkatesh J, Upadhyaya CP, Nookaraju A, Pandey SK, Park SW . Plant disease resistance genes: current status and future directions. Physiol Mol Plant Pathol 2012; 78: 51–65.
Lorang J, Kidarsa T, Bradford CS et al. Tricking the guard: exploiting plant defense for disease susceptibility. Science 2012; 338: 659–662.
Zhao H, Dong J, Wang T . Function and expression analysis of gibberellin oxidases in apple. Plant Mol Biol Report 2010; 28: 231–238.
Yang X, Brown SK, Davies PJ . The content and in vivo metabolism of gibberellin in apple vegetative tissues. J Am Soc Hortic Sci 2013; 138: 173–183.
Chiang HH, Hwang I, Goodman HM . Isolation of the Arabidopsis GA4 locus. The Plant Cell Online 1995; 7: 195–201.
Williams J, Phillips AL, Gaskin P, Hedden P . Function and substrate specificity of the gibberellin 3β-hydroxylase encoded by the Arabidopsis GA4 Gene. Plant Physiol 1998; 117: 559–563.
Mitchum MG, Yamaguchi S, Hanada A et al. Distinct and overlapping roles of two gibberellin 3-oxidases in Arabidopsis development. The Plant Journal 2006; 45: 804–818.
Lee S, Cheng H, King KE et al. Gibberellin regulates Arabidopsis seed germination via RGL2, a GAI/RGA-like gene whose expression is up-regulated following imbibition. Genes Dev 2002; 16: 646–658.
Cheng H, Qin L, Lee S et al. Gibberellin regulates Arabidopsis floral development via suppression of DELLA protein function. Development 2004; 131: 1055–1064.
Achard P, Renou JP, Berthomé R, Harberd NP, Genschik P . Plant DELLAs restrain growth and promote survival of adversity by reducing the levels of reactive oxygen species. Curr Biol 2008; 18: 656–660.
Rameau C . Strigolactones, a novel class of plant hormone controlling shoot branching. CR Biol 2010; 333: 344–349.
Foster TM, Watson AE, van Hooijdonk BM, Schaffer RJ . Key flowering genes including FT-like genes are upregulated in the vasculature of apple dwarfing rootstocks. Tree Genet Genomes 2014; 10: 189–202.
Loescher WH, Marlow GC, Kennedy RA . Sorbitol metabolism and sink-source interconversions in developing apple leaves. Plant Physiol 1982; 70: 335–339.
Etienne C, Rothan C, Moing A et al. Candidate genes and QTLs for sugar and organic acid content in peach [Prunus persica (L.) Batsch]. Theor Appl Genet 2002; 105: 145–159.
Newcomb RD, Crowhurst RN, Gleave AP et al. Analyses of expressed sequence tags from apple. Plant Physiol 2006; 141: 147–166.
Barratt DP, Derbyshire P, Findlay K et al. Normal growth of Arabidopsis requires cytosolic invertase but not sucrose synthase. Proc Natl Acad Sci USA 2009; 106: 13124–13129.
Tronconi MA, Fahnenstich H, Weehler MC et al. Arabidopsis NAD-malic enzyme functions as a homodimer and heterodimer and has a major impact on nocturnal metabolism. Plant Physiol 2008; 146: 1540–1552.
Acknowledgements
We gratefully acknowledge the Jiangsu Province Science Technology Independent Innovation Fund (CX (13) 3006) and the Sanxin Project of Jiangsu Province (SXGC (2013) 046) for providing financial support. We also acknowledge the National Natural Science Foundation of China (31171935) for providing financial support for a portion of this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Zhang, S., Chen, W., Xin, L. et al. Genomic variants of genes associated with three horticultural traits in apple revealed by genome re-sequencing. Hortic Res 1, 14045 (2014). https://doi.org/10.1038/hortres.2014.45
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hortres.2014.45
This article is cited by
-
MdWRKY75e enhances resistance to Alternaria alternata in Malus domestica
Horticulture Research (2021)
-
Identification and characterization of SNPs in released, landrace and wild accessions of mungbean (Vigna radiata (L.) Wilczek) using whole genome re-sequencing
Journal of Crop Science and Biotechnology (2021)
-
Genomic variation between PRSV resistant transgenic SunUp and its progenitor cultivar Sunset
BMC Genomics (2020)
-
Transcriptional regulation of bark freezing tolerance in apple (Malus domestica Borkh.)
Horticulture Research (2020)
-
Genetic relationships and population structure of pears (Pyrus spp.) assessed with genome-wide SNPs detected by genotyping-by-sequencing
Horticulture, Environment, and Biotechnology (2019)
