
Abstract
Biallelic mutations in the post-GPI attachment to proteins 3 (PGAP3) gene cause hyperphosphatasia with mental retardation syndrome 4 (HPMRS4), which is characterized by elevated serum alkaline phosphatase, severe psychomotor developmental delay, seizures, and facial dysmorphism. To date, 15 PGAP3 mutations have been reported in humans. Here we report a novel homozygous PGAP3 mutation (c.314C>A, p.Pro105Gln) in a Croatian patient and fully describe the clinical features.
Similar content being viewed by others


Hyperphosphatasia with mental retardation syndrome (HPMRS), also known as Mabry syndrome, is an autosomal recessive disease that is associated with inherited glycosylphosphatidylinositol (GPI) deficiencies (IGDs).1 There are two types of genes involved in IGDs: phosphatidylinositol glycan (PIG) genes and post-GPI attachment to proteins (PGAP) genes. PIG genes are involved in the biosynthesis and transfer of GPI, while PGAP genes play a role in the remodeling, sorting, and transport of GPI-anchored proteins (GPI-APs).1 To date, at least six genes responsible for HPMRS have been reported: PIGO (MIM 614730),2 PIGV (MIM 610274),3 PIGW (MIM 610275),4 PIGY (MIM 610662),5 PGAP2 (MIM 615187),6 and PGAP3 (MIM 611801, NM_033419.4).7 HPMRS is registered in the Online Mendelian Inheritance in Man (OMIM) database as a phenotypic series (HPMRS1-6, OMIM PS239300), and PGAP3 is the gene responsible for HPMRS4 (MIM 615716). Although there are a variety of clinical features of HPMRS, the major features include intellectual disability, hyperphosphatasia, seizures, and facial dysmorphism, as well as anomalies, such as brachytelephalangy.1,8
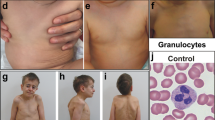
We encountered a patient with HPMRS. He was born to healthy and nonconsanguineous parents (from the same region) after an uneventful pregnancy with no asphyxia at birth (Apgar score was 10/10) (Figure 1a). He has no siblings, and his family history is unremarkable. His birth length, weight, and head circumference were 51 cm (+0.59 SD), 3,360 g (+0.03 SD), and 35.5 cm (+0.82 SD), respectively. Multiple congenital anomalies, including a broad nasal bridge, tented upper lip vermilion, cleft palate, low set ears, micrognathia, retrognathia, brachytelephalangy, left sided cryptorchidism, wide feet, and broad toes, were noticed at birth. Palatoplasty was performed at 1 year of age, and left orchiopexy was performed at 4 years of age. Progressive thoracic scoliosis started at 5 years of age and was treated with orthotics at 6 years with no improvements. At 2.5 months, he started intensive physical therapy for his marked generalized hypotonia that has been continued until now. He was unable to sit up by himself until 4 years of age. He first experienced recurrent generalized seizures with dysrhythmic electroencephalographic paroxysmal changes at 3 years of age, and these seizures were well controlled with phenobarbital. The seizures started again at 5 years of age and were controlled with valproate. When HPMRS was suspected at 6.5 years of age, he started treatment with 200 mg of pyridoxine daily and has been free of seizures since. Sleep disturbance (awake at night and sleeping during the daytime) started at 3 years of age, a condition that was normalized with pyridoxine. Pyridoxine was administered since it has been reported to be effective for neuronal symptoms in hyperphosphatasia with neuronal deficits.9 A brain magnetic resonance imaging at 5 years of age showed a thin corpus callosum without any other gross abnormalities (Figure 2).

Genetic analysis of the patient. (a) Familial pedigree and the PGAP3 mutation. (b) Human PGAP3 cDNA and the PGAP3 protein structure with previously reported mutations and a newly identified mutation in this study (underlined and in red). PGAP3 consists of eight exons, and the PGAP3 protein contains seven transmembrane domains (TMD). The description of the mutation was based on NM_033419.4. (c) Electropherograms of the patient (II-1) and his parents (I-1 and I-2). (d) Evolutionary conservation of p.Pro105 in PGAP3. Protein sequences of different species were aligned using CLUSTALW (http://www.genome.jp/tools/clustalw/).

Brain magnetic resonance imaging of the proband at the age of 5 years. T1- (a) and T2-weighted (b) axial images and a proton density-weighted sagittal image (c) are shown. A thin corpus callosum was observed (white arrows).
Currently, at the age of 8 years, he shows severe psychomotor developmental delay, autistic behavior, and bruxism. Eye contact is possible for short periods of time, but he has a short attention span. He can sit alone, crawl clumsily, and occasionally stand with support, but he cannot walk independently or speak any meaningful words. He often smiles and vocalizes with simple words. He cannot understand simple commands, but he can understand daily tasks, such as going for a walk or eating meals. He partially participates in dressing himself. He is unable to use cutlery and eats with his fingers. His vision and hearing are normal, but he cannot control egestion. His serum alkaline phosphatase (ALP) has been elevated (in the range from 500–1200 U/L) since the age of 1 year when it was first measured, and his ALP level was 578 U/L at the age of 7.5 years (reference range: 172–405 U/L). His serum 25-OH-vitamin-D3, calcium, and phosphorus levels have been in the normal range.
To identify the genetic cause of his condition, we performed whole-exome sequencing (WES) on the proband as reported previously.10 DNA was extracted from peripheral blood leukocytes after obtaining written informed consent. This study protocol was approved by the institutional review board of Yokohama City University School of Medicine. Using WES, 96.8% of the coding region of RefSeq genes was covered by 10 reads. We checked for variants in the known disease genes responsible for HPMRS and found a homozygous missense variant (NM_033419.4, c.314C>A, p.Pro105Gln) in PGAP3. This mutation was predicted as pathogenic by the in silico software: SIFT=0, PolyPhen2=1, and MutationTaster=disease causing. This variant was not registered in the Exome Aggregation Consortium (ExAC: http://exac.broadinstitute.org/), NHLBI GO Exome Sequencing Project 6500 (ESP6500: http://evs.gs.washington.edu/EVS/), Human Genetic Variation Database (HGVD: http://www.hgvd.genome.med.kyoto-u.ac.jp/), or our in-house exome database (n=575). Of note, this missense mutation was located at an identical position to a previously reported pathogenic mutation but led to a different base alteration to a different amino acid (c.314C>G, p.Pro105Arg) (Figure 1b).7 Sanger sequencing revealed that this new mutation was inherited from both unaffected carrier parents (Figure 1c). This amino residue is evolutionally conserved from C. elegans to H. sapiens (Figure 1d). In addition, similar to previously reported patients, the proband in this report showed facial dysmorphism, cryptorchidism, severe psychomotor developmental delay, speech impairment, hypotonia, autistic behavior, bruxism, and sleep disturbance (see also Supplementary Table S1).
To date, 15 mutations in PGAP3 have been reported in 30 patients with HPMRS 4 (Figure 1b).7,8,11–15 More than half of the mutations (9/15; 60%) are in exon 3 and exon 7, and these regions are considered hotspots for mutations.13,14 The mutation identified in our proband is also located within this hotspot in exon 3.
The clinical features of the current patient were compared with those in previously reported patients (Supplementary Table S1). All patients, including the one in this study, showed facial dysmorphism, motor delay, speech impairment, and elevated serum ALP. Interestingly, our patient also showed brachytelephalangy, one of the characteristic features of HPMRS. However, while brachytelephalangy has never been reported in HPMRS4 caused by the PGAP3 mutation, broad fingers and toenails have been reported in some patients with the PGAP3 mutation.7,8,11–15 This finding indicates that the PGAP3 mutation can cause brachytelephalangy, as the other types of HPMRS.
The proband in this study showed a thin corpus callosum, which is the most common type of brain abnormality among patients with the PGAP3 mutation. Interestingly, a thin corpus callosum is observed in all patients with a missense mutation (c.314C>G, p.Pro105Arg) or truncating mutation (c.402dupC, p.Met135Hisfs*28) in a homozygous state but not in those with other mutations. Some genotype–phenotype correlation is possible, although information is limited.
Hyperphosphatasia is an important clinical feature of HPMRS. All patients showed hyperphosphatasia (28/28; 100%), and most patients with PGAP3 mutations (22/28; 79%) had a higher serum ALP (1.1–3.6 times the upper limit) than the age-adjusted normal ranges (Supplementary Table S1). Patients with PIGO, PIGW, or PIGY mutations have been reported to have normal levels or mildly elevated ALP levels.5,13,16–18 By contrast, more than half of patients with PIGV or PGAP2 mutations showed extremely high levels of ALP (over 3.6 times the normal upper limit).6,17,19,20 The degree of elevated serum ALP may be related to the mutated genes.
In conclusion, we report HPMRS4 in a patient with a novel missense PGAP3 mutation. As there is a minimal number of patients with PGAP3 mutations, further analysis of these patients is required to understand the genotype–phenotype correlation.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Kinoshita T . Biosynthesis and deficiencies of glycosylphosphatidylinositol. Proc Jpn Acad Ser B Phys Biol Sci 2014; 90: 130–143.
Krawitz PM, Murakami Y, Hecht J, Kruger U, Holder SE, Mortier GR et al. Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am J Hum Genet 2012; 91: 146–151.
Krawitz PM, Schweiger MR, Rodelsperger C, Marcelis C, Kolsch U, Meisel C et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet 2010; 42: 827–829.
Maydan G, Noyman I, Har-Zahav A, Neriah ZB, Pasmanik-Chor M, Yeheskel A et al. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet 2011; 48: 383–389.
Ilkovski B, Pagnamenta AT, O'Grady GL, Kinoshita T, Howard MF, Lek M et al. Mutations in PIGY: expanding the phenotype of inherited glycosylphosphatidylinositol deficiencies. Hum Mol Genet 2015; 24: 6146–6159.
Hansen L, Tawamie H, Murakami Y, Mang Y, ur Rehman S, Buchert R et al. Hypomorphic mutations in PGAP2, encoding a GPI-anchor-remodeling protein, cause autosomal-recessive intellectual disability. Am J Hum Genet 2013; 92: 575–583.
Howard MF, Murakami Y, Pagnamenta AT, Daumer-Haas C, Fischer B, Hecht J et al. Mutations in PGAP3 impair GPI-anchor maturation, causing a subtype of hyperphosphatasia with mental retardation. Am J Hum Genet 2014; 94: 278–287.
Knaus A, Awaya T, Helbig I, Afawi Z, Pendziwiat M, Abu-Rachma J et al. Rare noncoding mutations extend the mutational spectrum in the PGAP3 subtype of hyperphosphatasia with mental retardation syndrome. Hum Mutat 2016; 37: 737–744.
Thompson MD, Killoran A, Percy ME, Nezarati M, Cole DE, Hwang PA . Hyperphosphatasia with neurologic deficit: a pyridoxine-responsive seizure disorder? Pediatr Neurol 2006; 34: 303–307.
Iwama K, Sasaki M, Hirabayashi S, Ohba C, Iwabuchi E, Miyatake S et al. Milder progressive cerebellar atrophy caused by biallelic SEPSECS mutations. J Hum Genet 2016; 61: 527–531.
Yavarna T, Al-Dewik N, Al-Mureikhi M, Ali R, Al-Mesaifri F, Mahmoud L et al. High diagnostic yield of clinical exome sequencing in Middle Eastern patients with Mendelian disorders. Hum Genet 2015; 134: 967–980.
Abouelhoda M, Sobahy T, El-Kalioby M, Patel N, Shamseldin H, Monies D et al. Clinical genomics can facilitate countrywide estimation of autosomal recessive disease burden. Genet Med 2016; 18: 1244–1249.
Pagnamenta AT, Murakami Y, Taylor JM, Anzilotti C, Howard MF, Miller V et al. Analysis of exome data for 4293 trios suggests GPI-anchor biogenesis defects are a rare cause of developmental disorders. Eur J Hum Genet 2017; 25: 669–679.
Abdel-Hamid MS, Issa MY, Otaify GA, Abdel-Ghafar SF, Elbendary HM, Zaki MS . PGAP3-related hyperphosphatasia with mental retardation syndrome: Report of 10 new patients and a homozygous founder mutation. Clin Genet 2017; 93: 84–91.
Nampoothiri S, Hebbar M, Roy AG, Kochumon SP, Bielas S, Shukla A et al. Hyperphosphatasia with mental retardation syndrome due to a novel mutation in PGAP3. J Pediatr Genet 2017; 6: 191–193.
Hogrebe M, Murakami Y, Wild M, Ahlmann M, Biskup S, Hortnagel K et al. A novel mutation in PIGW causes glycosylphosphatidylinositol deficiency without hyperphosphatasia. Am J Med Genet A 2016; 170: 3319–3322.
Xue J, Li H, Zhang Y, Yang Z. Clinical and genetic analysis of two Chinese infants with Mabry syndrome. Brain Dev 2016; 38: 807–818.
Zehavi Y, von Renesse A, Daniel-Spiegel E, Sapir Y, Zalman L, Chervinsky I et al. A homozygous PIGO mutation associated with severe infantile epileptic encephalopathy and corpus callosum hypoplasia, but normal alkaline phosphatase levels. Metab Brain Dis 2017; 32: 2131–2137.
Krawitz PM, Murakami Y, Riess A, Hietala M, Kruger U, Zhu N et al. PGAP2 mutations, affecting the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation syndrome. Am J Hum Genet 2013; 92: 584–589.
Jezela-Stanek A, Ciara E, Piekutowska-Abramczuk D, Trubicka J, Jurkiewicz E, Rokicki D et al. Congenital disorder of glycosylphosphatidylinositol (GPI)-anchor biosynthesis--the phenotype of two patients with novel mutations in the PIGN and PGAP2 genes. Eur J Paediatr Neurol 2016; 20: 462–473.
Data Citations
Matsumoto, Naomichi HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.1755 (2018)
Acknowledgements
We thank the patient and his family for their participation in this study. This work was supported by grants from Research on Measures for Intractable Diseases; Comprehensive Research on Disability Health and Welfare; the Strategic Research Program for Brain Science (SRPBS); the Practical Research Project for Rare/Intractable Diseases; the Initiative on Rare and Undiagnosed Diseases from the Japan Agency for Medical Research and Development; a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan; Grants-in-Aid for Scientific Research (A and B); Grant-in-Aid for Young Scientists (B); Challenging Exploratory Research from the Japan Society for the Promotion of Science; the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems from the Japan Science and Technology Agency; grants from the Ministry of Health, Labour and Welfare; and the Takeda Science Foundation. We thank Rhonna Gurevich, PhD, from the Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Sakaguchi, T., Žigman, T., Petković Ramadža, D. et al. A novel PGAP3 mutation in a Croatian boy with brachytelephalangy and a thin corpus callosum. Hum Genome Var 5, 18005 (2018). https://doi.org/10.1038/hgv.2018.5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2018.5
