
Abstract
Osteogenesis imperfecta (OI) is a heterogeneous disorder that is characterized by bone fragility and systemic complications, and is mainly caused by gene mutations in COL1A1 or COL1A2. A novel COL1A1 splicing mutation, c.750+2T>A, was identified in a Japanese OI family. Only the proband in this family showed various complications, such as heart valve diseases and severe scoliosis. The clinical heterogeneity in the family is discussed in this study.
Similar content being viewed by others

Osteogenesis imperfecta (OI) is a heterogeneous disorder that is characterized by bone fragility and systemic complications, and has a broad spectrum ranging from an asymptomatic or very mild form to a severe, lethal form.1,2 Generally, patients with OI show a normal intellectual ability and no neurological symptoms except for cases with complications due to brain nerves injuries resulting from basal skull bone compression.3 Recently, we identified a novel COL1A1 splicing mutation in a patient with OI phenotypes associated with various complications. Although his mother shared the same mutation, her OI phenotypes were not as severe. The clinical heterogeneity in the family is discussed in this study.
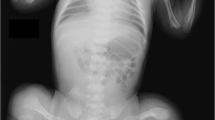
The proband is an adult 49-year-old male (III-7 in Figure 1a). He had a recurrent history of broken long bones (~30 episodes of bone fractures before adolescence), leading to secondary deformities in the bones and joints of his extremities. He underwent surgical operations, including a mitral valve repair and an aortic valve replacement associated with a coronary artery bypass graft at 32 and 36 years of age, respectively. The main four OI groups each display different modes of inheritance, with autosomal dominance being the predominant mode of inheritance in groups I and IV, and at least some families show autosomal recessive inheritance in OI types II and III, indicating genetic heterogeneity in OI. In the years following, the discovery of the COL1A1/2 mutations in all OI types, the four OI types were often used in clinical practice to categorize the severity of OI as mild and blue sclera (OI type 1), lethal (OI type 2), severely deforming (OI type 3), and moderately deforming with normal sclera (OI type 4).4,5 The patient had blue sclera, a short stature, mild dentinogenesis imperfecta, and severe scoliosis (Figure 1b). Based on these findings, he was clinically diagnosed with OI type I.4–6 Currently, he works as a pediatrician.

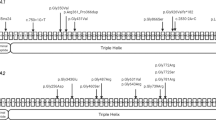
Clinical information and the results of the molecular analysis. (a) The family tree of the proband’s relatives. The proband (III-7) is a patient with OI. His grandfather (I-1), mother (II-5), and aunt (II-1) are suspected to have OI based on their clinical features, including a short stature, a history of long bone fractures, and blue sclera, although they did not exhibit scoliosis or heart disease. Causes of death: (I-1) death in action, (I-2) blood disease at 84 years, (II-1) traffic accident at 72 years, (III-2) heart disease at 25 years, (III-4) accident at 18 years. (b) Severe scoliosis (Cobb angle: right ~70° at T5-T10, left 60° at T11-L3). There is no obvious progression after adolescence. (c) The IGV (http://www.broadinstitute.org/igv/) shows the identified COL1A1 variant in ~50% of the reads. (d) Electropherograms of Sanger sequencing. The heterozygous variant in the consensus sequence in the splicing donor site of intron 10 is shown in the proband and his mother. The wild-type sequence is observed in the father. IGV, Integrative Genomics Viewer; OI, osteogenesis imperfecta.
He had a family history (Figure 1a). His grandfather (I-1), mother (II-5), and aunt (II-1) also had a short stature, blue sclera, and histories of recurrent long bone fractures but no deformities in the long bones. These family histories suggested autosomal dominant trait disorders, which are compatible with OI type I.5,6 However, there was intra-familial variability. The proband showed more remarkable and serious clinical features, i.e., more frequent bone fractures, severe bone deformities, severe scoliosis, and a complication of heart defects (Table 1). In comparison, his mother and aunt showed similar, less severe manifestations. Because the aunt died due to a traffic accident, detailed information is unavailable. To elucidate the clinical variability, a molecular analysis was performed in this family.
Although it has been revealed that many genes are related to OI, most OI cases are caused by mutations in the genes encoding collagen type I alpha-1 and -2 chains (COL1A1 and COL1A2).3 Thus, these two genes were proposed as candidate targets. COL1A1 and COL1A2 are too large to analyze by Sanger sequencing, and next generation sequencing (NGS) was used as the analysis method.7 The study was performed in accordance with the Helsinki Declaration. We obtained permission from the ethical committee of the institution. The patients were enrolled in this study after obtaining written informed consent through careful genetic counseling regarding the appropriate handling of genetic information and the possible incidental findings.
Blood samples were obtained from the proband and his parents. DNA was then extracted using the QIAamp DNA extraction kit (QIAGEN, Hilden, Germany). NGS using TruSight One v1.0 sequencing panel (Illumina, San Diego, CA, USA) for the proband was performed first to screen single-nucleotide variants, as described previously.8 The mean coverage depth was 95.7×. The extracted data were mapped to a reference genome (GRCh37/hg19) using BWA Enrichment v1.0 cloud software (Illumina) and annotated using Variant Studio software (Illumina). As a result, 8040 variants were obtained. A routine filtering was performed, and a heterozygous variant at the splicing donor site, NM_000088.3(COL1A1):c.750+2T>A, was identified (Figure 1c). There were no possible variants in the other OI related genes, including BMP1, COL1A2, CRTAP, FGFR2, FGFR3, FKBP10, LEPRE1, LRP5, PPIB, SERPINF1, SERPINH1, and SP7.
The identified variant was re-confirmed by standard Sanger sequencing as described previously.8 The parental samples were also assessed, and the mother was confirmed to be a carrier of this variant, as suspected (Figure 1d). Because the identified variant, c.750+2T>A, is located on the consensus sequence of the intron 10 splicing donor site, it was suspected to lead to a splicing abnormality. Although the same variant has never been reported previously or registered in the Database of Collagen Mutations (http://www.le.ac.uk/genetics/collagen/),9,10 similar splicing donor site variants were frequently identified in OI patients. Therefore, the identified variant was considered a causative mutation.
The clinical features observed in the proband are serious in comparison with those of the other family members. It may be due to clinical variability. Although significant phenotypic differences within one family are rare, there are some reports of intra-familial variability.11 Furthermore, it is known that the clinical severity is not exactly the same between OI patients with the same mutation from unrelated families.12 Somatic mosaicism has often been reported as an explanation of intra-familial variability in OI phenotypes.13 However, the family members in three generations were hypothesized to have the same condition in the present study, and this inheritance pattern is inconsistent with the finding of somatic mosaicism.
One of the possible reasons for the intra-familial variability in this study may be the patient’s lifestyle.12 The proband reported his active lifestyle as follows: a traumatic accident by slipping on a rainy day; increasing the range of his activities; and carrying or lifting heavy things, such as books or other weights, in addition to participating in exercise such as swimming for 15 years since his school days. An unidentifiable genetic modifier may be another reason.
In addition to these activities, weight gain and a diet lacking in protein, vitamins and calcium might exacerbate bone fractures in OI patients. Connective tissues that are too flexible and systemic muscles that are too thin to support bones may also accelerate the development of valve regurgitation and scoliosis. The recognition of the natural history of OI patients is important for supporting their quality of life.14 It is predicted that if we know how to avoid these risks, we may be able to improve the disease prognosis. In the future, it is necessary to elucidate the exact elements needed for a better quality of life. Therefore, we should collect long natural histories of OI patients, including possible factors that may affect the prognosis, in association with the genotypic data.
References
References
Shapiro JR, Byers PH, Glorieux FH, Sponseller P (eds). Osteogenesis Imperfecta: A Translational Approach to Brittle Bone Disease. Academic Press: Cambridge, MA: 2013.
Takagi M, Matsushita M, Nishimura G, Hasegawa T . Osteogenesis imperfecta IIC caused by a novel heterozygous mutation in the C-propeptide region of COL1A1. Hum Genome Var 2014; 1: 14025.
Forlino A, Marini JC . Osteogenesis imperfecta. Lancet 2016; 387: 1657–1671.
Sillence DO, Senn A, Danks DM . Genetic heterogeneity in osteogenesis imperfecta. J Med Genet 1979; 16: 101–116.
Van Dijk FS, Sillence DO . Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A 2014; 164A: 1470–1481.
Van Dijk FS, Pals G, Van Rijn RR, Nikkels PG, Cobben JM . Classification of osteogenesis imperfecta revisited. Eur J Med Genet 2010; 53: 1–5.
Arvai K, Horvath P, Balla B, Tobias B, Kato K, Kirschner G et al. Next-generation sequencing of common osteogenesis imperfecta-related genes in clinical practice. Sci Rep 2016; 6: 28417.
Yamamoto T, Shimojima K . A novel MED12 mutation associated with non-specific X-linked intellectual disability. Hum Genome Var 2015; 2: 15018.
Dalgleish R . The human type I collagen mutation database. Nucleic Acids Res 1997; 25: 181–187.
Dalgleish R . The Human Collagen Mutation Database 1998. Nucleic Acids Res 1998; 26: 253–255.
Moraes MV, Milanez M, Almada BV, Sipolatti V, Reboucas MR, Nunes VR et al. Variable expressivity of osteogenesis imperfecta in a Brazilian family due to p.G1079S mutation in the COL1A1 gene. Genet Mol Res 2012; 11: 3246–3255.
Liu HY, Huang J, Wu D, Li T, Guo LJ, Guo QN et al. Collagen type I alpha 1 mutation causes osteogenesis imperfecta from mild to perinatal death in a chinese family. Chin Med J (Engl) 2016; 129: 88–91.
Cohen-Solal L, Zylberberg L, Sangalli A, Gomez Lira M, Mottes M . Substitution of an aspartic acid for glycine 700 in the alpha 2(I) chain of type I collagen in a recurrent lethal type II osteogenesis imperfecta dramatically affects the mineralization of bone. J Biol Chem. 1994; 269: 14751–14758.
Tsimicalis A, Denis-Larocque G, Michalovic A, Lepage C, Williams K, Yao TR et al. The psychosocial experience of individuals living with osteogenesis imperfecta: a mixed-methods systematic review. Qual Life Res 2016; 25: 1877–1896.
Data Citations
Seto, Toshiyuki HGV Database (2017) http://dx.doi.org/10.6084/m9.figshare.hgv.950
Acknowledgements
We would like to express our gratitude to the patient and his parents for their cooperation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Seto, T., Yamamoto, T., Shimojima, K. et al. A novel COL1A1 mutation in a family with osteogenesis imperfecta associated with phenotypic variabilities. Hum Genome Var 4, 17007 (2017). https://doi.org/10.1038/hgv.2017.7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2017.7
