
Abstract
The mitochondrial aspartyl-tRNA synthetase 2 gene (DARS2) is responsible for leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL). A Japanese patient with LBSL showed compound heterozygous DARS2 mutations c.358_359delinsTC (p.Gly120Ser) and c.228-15C>G (splicing error). This provides further evidence that most patients with LBSL show compound heterozygous mutations in DARS2 in association with a common splicing mutation in the splicing acceptor site of intron 2.
Similar content being viewed by others
Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL, MIM#611105) is an autosomal recessive disorder of the brain white matter and clinically characterized by slowly progressive pyramidal, cerebellar, and dorsal column dysfunction involving the legs more than the arms.1 Distinct radiological findings include heterogeneous cerebral white matter abnormalities accompanied by a selective involvement of the brainstem and spinal cord tracts. Elevated lactate levels in magnetic resonance spectroscopy of the abnormal white matter is also characteristic. It has been reported that the clinical spectrum may vary from early-onset severe disease, which is sometimes even fatal within the first few years, to an adult-onset type.1 Variable clinical manifestations, including delayed intellectual and/or motor development, cognitive impairment, epilepsy, and peripheral neuropathy, may also occur. Definite diagnosis of LBSL is established by the demonstration of mutations in the mitochondrial aspartyl-tRNA synthetase 2 gene (DARS2), which encodes mitochondrial aspartyl-tRNA synthetase, the enzyme that attaches the amino acid aspartate to the correct mitochondrial transfer RNA.2 Aspartyl-tRNA is necessary in the translation of mitochondrial messenger RNA into protein.3 The majority of the LBSL cases is due to compound heterozygous DARS2 mutations.
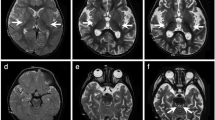
Here we present a male with LBSL in whom a novel DARS2 mutation was identified. At present, the patient is 27 years of age. He is the second child of healthy and non-consanguineous Japanese parents after an uneventful pregnancy and delivery. He has two healthy siblings. He started to walk at 1 year and 8 months. It was noted that he easily fell to the ground when he was 3.5 years. At that time, brain computed tomography was performed, and leukoencephalopathy was suggested. From the age of 4 years, he started to require support for walking. A walking aid and a wheelchair were needed from the age of 6 years and 10 years, respectively. Subsequently, nystagmus was noted at 13 years. His motor ability gradually deteriorated and he, at present, can use only his upper limbs. Intellectual ability has also declined. Routine laboratory examination, including metabolic screening and cerebrospinal fluid testing, showed no abnormalities involving lactic and pyruvate acids. Enzymatic analyses for arylsulfatase A and galactocerebrosidase were also normal. Brain magnetic resonance imaging examined at 27 years revealed white matter abnormalities in association with cystic findings (Figures 1a–d). The spinal cord was also involved. These findings were typical for LBSL. Although an elevation of lactate level was not clear by magnetic resonance spectroscopy at the same time (data not shown),4,5 such a pattern is not unusual. There are some LBSL patients who show normal lactate levels, which might occur due to advanced stage or a late onset of symptoms.6–8

Radiological findings and results of molecular analysis. (a–d) Axial magnetic resonance images examined at 27 years. T2-weighted axial images (a–c) and a fluid-attenuated inversion recovery image (d). (a) In the spinal cord, the posterior columns (yellow) and the lateral corticospinal tracts (red) are involved. (b) At the level of the pons, high signal intensity is shown in bilateral lesions in the pyramidal tracts (red), medial lemniscus (yellow), superior cerebellar peduncles (blue), and intraparenchymal trajectory of the trigeminal nerve (green). (c) At the level of the basal ganglia, the posterior limb of the internal capsule and periventricular white matter are affected. (d) High signal intensity is shown in the periventricular white matter associated with multiple small cysts. (e, f) Electropherograms of Sanger sequencing for the patient, his mother, and the sister. Although a c.228-20T>C polymorphism is shown in both the patient and his sister (e), c.228-15C>G is only present in the patient. c.358_359delinsTC (p.Gly120Ser) is commonly shown in all samples (f), and the affected amino acid is conserved among species (g).
This study was performed in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of Tokyo Women’s Medical University. After obtaining written informed consent, blood samples were obtained from the patient, his mother, and his younger sister. The father’s sample was not obtained, because the couple was divorced. Although LBSL was suspected based on the abovementioned findings, whole-exome sequencing was performed. Briefly, DNA samples were captured using the SureSelect Human All Exon Kit v5 (Agilent Technologies, Santa Clara, CA) and sequenced on an Illumina HiSeq 2500 system (Illumina, San Diego, CA) with 101 bp paired-end reads. Exome data processing, variant calling, and variant annotation were performed as previously described.9 The average read depth of protein-coding regions ranged from 87.29 to 118.99×(mean coverage against coding regions: 118.99×(proband), mother: 87.29×(mother), 87.76×(sister)), and at least 91.2% of target bases were sequenced with 10 or more reads for each sample (96.5% (proband), 91.2% (mother), and 92.5% (sister)). Common single-nucleotide polymorphisms with minor allele frequencies ⩾1% in dbSNP 135 and variants observed in more than five subjects of our in-house exome database (n=575 controls) were filtered out. We evaluated the remaining variants using SIFT (http://sift.jcvi.org/) and Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/) prediction software. Particular attention was given to mutations found in genes known to cause neurodegenerative diseases when mutated. The samples from the mother and the sister were used for filtering. Finally, two heterozygous mutations in DARS2, NM_018122.4: c.228-15C>G and c.358_359delinsTC (p.Gly120Ser) remained, and mutations in the other candidate genes for leukoencephalopathy were ruled out. p.Gly120Ser was detected in the mother and healthy sister (Figures 1e and f). Although p.Gly120Ser has never been reported previously (Supplementary Table S1), the affected codon is conserved among species (Figure 1g) and prediction scores including SIFT and Polyphen-2 suggested that this mutation is ‘damaging.’ Therefore, we concluded that this mutation is pathogenic. On the other hand, the other mutation, c.228-15C>G, has been reported previously as a pathogenic splicing mutation that causes predisposition to p.R76SfsX5. As this splicing mutation was not detected in the mother and healthy sister, this was suspected to be inherited from his father or to be a de novo mutation. From these findings, we concluded that LBSL features in this patient are derived from compound heterozygous mutations in DARS2.
In 2007, Scheper et al.2 reported that mutations in the DARS2 gene are responsible for this disease. Since then, more than 50 types of DARS2 mutations have been identified.3,4,6,8,10–19 It is striking that most patients have been reported to have compound heterozygous mutations in DARS2 and that one of the mutations is often located on the splicing acceptor site in intron 2 (the region neighboring exon 3).2,3 Because the patient showed the same pattern, this report suggests additional evidence supporting this finding.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
van der Knaap MS, van der Voorn P, Barkhof F, Van Coster R, Krageloh-Mann I, Feigenbaum A et al. A new leukoencephalopathy with brainstem and spinal cord involvement and high lactate. Ann Neurol 2003; 53: 252–258.
Scheper GC, van der Klok T, van Andel RJ, van Berkel CG, Sissler M, Smet J et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet 2007; 39: 534–539.
van Berge L, Hamilton EM, Linnankivi T, Uziel G, Steenweg ME, Isohanni P et al. Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation: clinical and genetic characterization and target for therapy. Brain 2014; 137: 1019–1029.
Schicks J, Schols L, van der Knaap MS, Synofzik M . Teaching NeuroImages: MRI guides genetics: leukoencephalopathy with brainstem and spinal cord involvement (LBSL). Neurology 2013; 80: e176–e177.
Yamashita S, Miyake N, Matsumoto N, Osaka H, Iai M, Aida N et al. Neuropathology of leukoencephalopathy with brainstem and spinal cord involvement and high lactate caused by a homozygous mutation of DARS2. Brain Dev 2013; 35: 312–316.
Lin J, Chiconelli Faria E, Da Rocha AJ, Rodrigues Masruha M, Pereira Vilanova LC, Scheper GC et al. Leukoencephalopathy with brainstem and spinal cord involvement and normal lactate: a new mutation in the DARS2 gene. J Child Neurol 2010; 25: 1425–1428.
Petzold GC, Bohner G, Klingebiel R, Amberger N, van der Knaap MS, Zschenderlein R . Adult onset leucoencephalopathy with brain stem and spinal cord involvement and normal lactate. J Neurol Neurosurg Psychiatry 2006; 77: 889–891.
Labauge P, Dorboz I, Eymard-Pierre E, Dereeper O, Boespflug-Tanguy O . Clinically asymptomatic adult patient with extensive LBSL MRI pattern and DARS2 mutations. J Neurol 2011; 258: 335–337.
Miyatake S, Okamoto N, Stark Z, Nabetani M, Tsurusaki Y, Nakashima M et al. ANKRD11 variants cause variable clinical features associated with KBG syndrome and Coffin-Siris-like syndrome. J Hum Genet 2017; 62: 741–746.
Uluc K, Baskan O, Yildirim KA, Ozsahin S, Koseoglu M, Isak B et al. Leukoencephalopathy with brain stem and spinal cord involvement and high lactate: a genetically proven case with distinct MRI findings. J Neurol Sci 2008; 273: 118–122.
Mikhaylova SV, Zakharova E, Banin AV, Demushkina AA, Petrukhin AS . Clinical and molecular genetic diagnosis of leuoencephalopathy with brainstem and spinal cord involvement and lactate elevation in children. Zh Nevrol Psikhiatr Im S S Korsakova 2009; 109: 16–22.
Sharma S, Sankhyan N, Kumar A, Scheper GC, van der Knaap MS, Gulati S . Leukoencephalopathy with brain stem and spinal cord involvement and high lactate: a genetically proven case without elevated white matter lactate. J Child Neurol 2011; 26: 773–776.
Miyake N, Yamashita S, Kurosawa K, Miyatake S, Tsurusaki Y, Doi H et al. A novel homozygous mutation of DARS2 may cause a severe LBSL variant. Clin Genet 2011; 80: 293–296.
Synofzik M, Schicks J, Lindig T, Biskup S, Schmidt T, Hansel J et al. Acetazolamide-responsive exercise-induced episodic ataxia associated with a novel homozygous DARS2 mutation. J Med Genet 2011; 48: 713–715.
Huang Q, Xiao J, Wang J, Jiang Y, Wu Y . Clinical and genetic analysis of a family with leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Zhongghua Er Ke Za Zhi 2012; 50: 50–55.
Tzoulis C, Tran GT, Gjerde IO, Aasly J, Neckelmann G, Rydland J et al. Leukoencephalopathy with brainstem and spinal cord involvement caused by a novel mutation in the DARS2 gene. J Neurol 2012; 259: 292–296.
Cheng FB, Shen PP, Zhou HW, Meng HM, Yang Y, Feng JC . Adult-onset leukoencephalopathy with brain stem and spinal cord involvement in Chinese Han population: a case report and literature review. Neurol India 2013; 61: 161–163.
Köhler C, Heyer C, Hoffjan S, Stemmler S, Lücke T, Thiels C et al. Early-onset leukoencephalopathy due to a homozygous missense mutation in the DARS2 gene. Mol Cell Probes 2015; 29: 319–322.
Lan MY, Chang YY, Yeh TH, Lin TK, Lu CS . Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL) with a novel DARS2 mutation and isolated progressive spastic paraparesis. J Neurol Sci 2017; 372: 229–231.
Data Citations
Yamamoto, Toshiyuki HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.1726 (2017)
Yamamoto, Toshiyuki HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.1729 (2017)
Acknowledgements
We express our gratitude to the patient and his family for their cooperation. This work is supported by a Research on Measures for Intractable Diseases grant (N Matsumoto), a Comprehensive Research on Disability Health and Welfare grant (N Matsumoto), the Strategic Research Program for Brain Science (N Matsumoto), and the Practical Research Project for Rare/Intractable Diseases (N Matsumoto and TY) from the Japanese Agency for Medical Research and Development (AMED); a grant-in-aid for Scientific Research on Innovative Areas (Transcription Cycle; N Miyake and N Matsumoto) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT); grants-in-aid for Scientific Research (A (N Matsumoto), B (N Miyake ), and C (SM and TY)); the Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems (N Matsumoto) from the Japan Science and Technology Agency (JST); and grants from the Ministry of Health, Labour and Welfare (N Matsumoto), The Ichiro Kanehara Foundation (SM), and the Takeda Science Foundation (N Miyake and N Matsumoto).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Human Genome Variation website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Shimojima, K., Higashiguchi, T., Kishimoto, K. et al. A novel DARS2 mutation in a Japanese patient with leukoencephalopathy with brainstem and spinal cord involvement but no lactate elevation. Hum Genome Var 4, 17051 (2017). https://doi.org/10.1038/hgv.2017.51
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2017.51
This article is cited by
-
Mitochondrial aminoacyl-tRNA synthetase disorders: an emerging group of developmental disorders of myelination
Journal of Neurodevelopmental Disorders (2019)
