
Abstract
The tubulin beta-4A gene (TUBB4A) is associated with two different clinical conditions, dystonia type 4 (DYT4) and hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC). We identified a novel TUBB4A mutation, c.286G>A (p.G96R), in an adult male patient who suffered neurological symptoms beyond adolescence. This patient shows intermediate clinical features between DYT4 and H-ABC, suggesting that the TUBB4A disorder would constitute a spectrum disorder.
Similar content being viewed by others

H-ABC (MIM#612438 (hypomyelinating leukodystrophy-6; HLD6)) is a rare leukodystrophy caused by mutations in the TUBB4A.1,2 H-ABC is characterized by clinical features associated with hypomyelination, i.e., delayed motor development, extrapyramidal movement disorders (dystonia, choreoathetosis, rigidity, opisthotonus and oculogyric crises), ataxia, progressive spastic tetraplegia, and in some cases, seizures. Patients showed clinical onset before 3 years of age.1,3 The final diagnosis of H-ABC is based on distinctive magnetic resonance imaging (MRI) findings, including the combination of hypomyelination, cerebellar atrophy and disappearance of the putamen.
TUBB4A mutations were also associated with autosomal dominant torsion DYT4 (MIM#128101).4 Patients with DYT4 exhibit normal MRI findings, but have a ‘whispering’ dysphonia, generalized dystonia and gait ataxia. Clinical onsets of DYT4 patients are in second to third decade of life. Although TUBB4A mutations have been identified in familial cases with DYT4, the severity of the clinical symptoms in H-ABC patients precludes offspring and all identified TUBB4A mutations in H-ABC patients were de novo, except for a definite mosaic inheritance.3
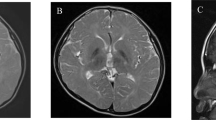
A 21-year-old Japanese male patient showed an unremarkable family history. His abnormal gait was first observed beyond teen age although definite onset time is ambigious. The patient was referred to a hospital when he was 17 years old. Upon physical examination, he showed no abnormalities or hepatosplenomegaly. However, neurological examination showed mild spasticity in his lower extremities. An ophthalmological examination showed a cherry-red spot that suggests atrophy of the optic nerve but no nystagmus. Brain MRI showed an abnormal intensity in the white matter (diffuse high intensity by T2 and fluid attenuated inversion recovery images; Figure 1a), but no atrophic findings in the basal ganglia and cerebellum were observed. An auditory brain response test showed a delayed peak of the wave V in both sides (left: 7.1 msec, right: 6.8 msec). Finally, visual-evoked potential and somatosensory-evoked potential tests of the upper extremities also showed delayed peaks.

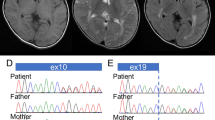
Clinical and molecular information. (a) A T2-weighted axial image of brain MRI indicates high intensity in the white matter. Slightly dilated lateral ventricles may indicate the mild atrophy of the basal ganglia. (b) Electropherograms of Sanger sequencing for the present family. Only the patient shows heterozygous mutation of c.286G>A. (c) Comparison of the amino-acid sequences with different species. The affected residue is conserved among species. (d) Mapping TUBB4A mutations to the 3-D protein crystal structure of the bovine αβ-tubulin heterodimer. The bovine α- and β-tubulins are shown in gray and green, respectively. In this image, β-tubulin is supposed to be encoded by TUBB4A whereas α-tubulin is uncertain. The guanosine triphosphate (GTP), Taxol and guanosine diphosphate (GDP) are marked with brown lines. The locations of the mutated residues are highlighted as yellow dot-halos. The residues associated with H-ABC are marked with bold black lines. The residues not associated with H-ABC but associated with other diseases, such as isolated hypomyelination, hypomyelinating leukodystrophy and spastic paraplegia, are marked with gray lines. The residue p.F367 is located behind Taxol in the figure. The location of the present mutation, p.G96R, is marked with a red line. The amino acid at the position 364 is Ala(A) in human but Ser(S) in bovine. GTP, guanosine triphosphate; H-ABC, hypomyelination with atrophy of the basal ganglia and cerebellum; MRI, magnetic resonance imaging.
When he was 19 years old, this patient started to show mildly dystonic movements. There was no dysphonia and dysarthria. His intelligence quotient was evaluated as 74 (language: 84, performance: 68) by the Wechsler Adult Intelligence Scale-III. Although the Mini-Mental State Examination showed a normal score of 27/30, the ‘Kana-hiroi’ test showed significantly lower scores in comparison to the age-matched mean score.5 Finally, a test for stabilometry revealed a horizontal postural sway with and without opening of the eyes. There was no remarkable abnormality in laboratory examination. At present, he still can play table tennis.
This study was performed in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of Tokyo Women’s Medical University. After receiving written informed consent, next-generation sequencing using the TruSight One v1.0 sequencing panel (Illumina, San Diego, CA, USA) was performed, as described.6 However, no possible candidate variants were detected. Since TUBB4A was not included in the panel, Sanger sequencing for all TUBB4A exons was then performed as described.7 As a result, NM_006087.3(TUBB4A):c.286G>A [p.Gly(G)96Arg(R)] was detected in the conserved region (Figures 1b and c). This was considered as de novo mutation because both parents did not carry this mutation. The prediction scores of this mutation, which was evaluated by wANNOVAR (http://wannovar.usc.edu/), suggested this mutation was ‘damaging’ (Supplementary Table S1). This mutation was not registered in the database including Human Genetic Variation Database (http://www.hgvd.genome.med.kyoto-u.ac.jp/), the Genome Aggregation Database (http://gnomad.broadinstitute.org/) and the Exome Aggregation Consortium (http://exac.broadinstitute.org/). Based on these findings, the c.286G>A [p.G96R] mutation was considered to be disease-causing.
The previously reported mutations in the coding regions of TUBB4A are summarized in Supplementary Table S2. A total of 36 mutations have been reported and did not include the mutation identified in this study, indicating that p.G96R is a novel disease-causing mutation. Furthermore, the ages of onset for the reported patients are also listed in Supplementary Table S2. Based on these data, the present patient was the oldest patient among those with hypomyelinating leukodystrophy.
Mutations in TUBB4A have been reported in association with two different disorders including DYT4, which is characterized by later adult-onset dystonia with normal brain MRI findings,4 and H-ABC, which is characterized by neonatal or earlier onset and traits of hypomyelination, cerebellar atrophy and progressive degeneration of the putamen and the caudate nucleus.1,2 Since both DYT4 and H-ABC are due to the same disease-causing gene and result in the neurological impairments, it remains unclear whether these two diseases are allelic diseases or two ends of a clinical spectrum.8 Indeed, severe generalized dystonia with subtle dysphonia was observed in patients with DYT4, while in some cases of H-ABC, less affected features (an intermediate clinical phenotype between DYT4 and H-ABC) were observed.8
A striking distinction between DYT4 and H-ABC can be revealed from different features present in the brain MRI. However, diverse extension of cerebellar atrophy has been observed in patients with H-ABC.3 Partial MRI features were also reported in some H-ABC patients, where the basal ganglia and cerebellum were still relatively preserved.8 In the present patient, an abnormal intensity in the white matter was observed but atrophy of the basal ganglia and cerebellum was not. The age onset of the present patient was extremely higher than that generally observed in patients with H-ABC (Supplementary Table S2).
Most of the patients with DYT4 start to show clinical features during the second to third decade of life. Because the present patient is 21 years old, it is reasonable that dystonic movements and a horizontal postural sway observed in the present patient are related to DYT4 features. From these findings, the present patient may be categorized as an intermediate type of the TUBB4A disorder. DYT4 and H-ABC would constitute the same spectrum disorders depending on the spatio-temporal dynamics of TUBB4A expressions.3,8,9
Previously, 36 different TUBB4A mutations have been reported.10,11,12,13,14,15,16,17,18,19 In order to investigate how these mutations affect the microtubule structure and why the different mutations lead to various clinical features, the FirstGlance in Jmol macromolecular visualization tool (http://bioinformatics.org/firstglance/fgij/) was used to process data from the Protein Data Bank.20 All of the previously reported TUBB4A mutations were mapped to the bovine αβ-tubulin protein crystal structure (Protein Data Bank identification code: 1JFF) which is similar to the human αβ-tubulin protein structure (Figure 1d).20 The most commonly reported H-ABC mutation is c.745G>A (p.D249N), which is located in the T7 loop of the protein. The residue D249 is close to the guanosine triphosphate domain and is speculated to play an important role in coordinating an interaction between the T7 loop and the α-tubulin-bound guanosine triphosphate. The other mutations close to the guanosine triphosphate domain, such as p.A352T, p.C354Y and p.M323R, cause the typical H-ABC phenotypes.3,8
Intriguingly, the mutations located on the outside of the αβ-tubulin heterodimer, which is distant from the guanosine triphosphate domain, are likely to result in milder phenotypes without atrophy of the basal ganglia and cerebellum. For example, the p.E410K mutation was reported in patients with spastic paraparesis and segmental dystonia,21 and the p.V180G and p.R391H mutations were related to isolated hypomyelination.9,22 One exception is a heterozygous mutation at residue p.R2. The mutation p.R2G is associated with DYT4 phenotypes, whereas p.R2Q and p.R2W result in H-ABC phenotypes. The possible explanation for this different clinical outcome arisen by the different amino-acid alteration at the same residue.
In conclusion, the c.286G>A (p.G96R) mutation identified in this study is a novel mutation located on the outside of the αβ-tubulin heterodimer. The patient exhibited hypomyelinating leukodystrophy that was associated with milder phenotypic features and a later age of onset. These findings are consistent with the proposed genotype-phenotype correlation. Overall, the neurological phenotype, the clinical features and the amino acid change of this mutation expand our current understanding of the clinical spectrum for TUBB4A disorder.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
van der Knaap MS, Naidu S, Pouwels PJ, Bonavita S, van Coster R, Lagae L et al. New syndrome characterized by hypomyelination with atrophy of the basal ganglia and cerebellum. AJNR Am J Neuroradiol 2002; 23: 1466–1474.
Simons C, Wolf NI, McNeil N, Caldovic L, Devaney JM, Takanohashi A et al. A de novo mutation in the beta-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet 2013; 92: 767–773.
Hamilton EM, Polder E, Vanderver A, Naidu S, Schiffmann R, Fisher K et al. Hypomyelination with atrophy of the basal ganglia and cerebellum: further delineation of the phenotype and genotype-phenotype correlation. Brain 2014; 137: 1921–1930.
Hersheson J, Mencacci NE, Davis M, MacDonald N, Trabzuni D, Ryten M et al. Mutations in the autoregulatory domain of β-tubulin 4a cause hereditary dystonia. Ann Neurol 2013; 73: 546–553.
Maeshima S, Funahashi K, Itakura T, Komai N, Dohi N . Computed topographic electroencephalographic study in left hemiplegic patients with higher cortical dysfunction. Arch Phys Med Rehabil 1994; 75: 189–192.
Shimojima K, Okamoto N, Yamamoto T . A novel TUBB3 mutation in a sporadic patient with asymmetric cortical dysplasia. Am J Med Genet A 2016; 170A: 1076–1079.
Shimojima K, Okumura A, Ikeno M, Nishimura A, Saito A, Saitsu H et al. A de novo TUBB4A mutation in a patient with hypomyelination mimicking Pelizaeus-Merzbacher disease. Brain Dev 2014; 37: 281–285.
Erro R, Hersheson J, Ganos C, Mencacci NE, Stamelou M, Batla A et al. H-ABC syndrome and DYT4: Variable expressivity or pleiotropy of TUBB4 mutations? Mov Disord 2015; 30: 828–833.
Pizzino A, Pierson TM, Guo Y, Helman G, Fortini S, Guerrero K et al. TUBB4A de novo mutations cause isolated hypomyelination. Neurology 2014; 83: 898–902.
Miyatake S, Osaka H, Shiina M, Sasaki M, Takanashi J, Haginoya K et al. Expanding the phenotypic spectrum of TUBB4A-associated hypomyelinating leukoencephalopathies. Neurology 2014; 82: 2230–2237.
Lohmann K, Wilcox RA, Winkler S, Ramirez A, Rakovic A, Park J-S et al. Whispering dysphonia (DYT4 dystonia) is caused by a mutation in the TUBB4 gene. Ann Neurol 2013; 73: 537–545.
Purnell SM, Bleyl SB, Bonkowsky JL . Clinical exome sequencing identifies a novel TUBB4A mutation in a child with static hypomyelinating leukodystrophy. Pediatr Neurol 2014; 50: 608–611.
Tonduti D, Aiello C, Renaldo F, Dorboz I, Saaman S, Rodriguez D et al. TUBB4A-related hypomyelinating leukodystrophy: New insights from a series of 12 patients. Eur J Paediatr Neurol 2016; 20: 323–330.
Isakov O, Lev D, Blumkin L, Celniker G, Leshinsky-Silver E, Shomron N . Crowdfunding effort identifies the causative mutation in a patient with nystagmus, microcephaly, dystonia and hypomyelination. J Genet Genomics 2015; 42: 79–81.
Kancheva D, Chamova T, Guergueltcheva V, Mitev V, Azmanov DN, Kalaydjieva L et al. Mosaic dominant TUBB4A mutation in an inbred family with complicated hereditary spastic paraplegia. Mov Disord 2015; 30: 854–858.
Arai-Ichinoi N, Uematsu M, Sato R, Suzuki T, Kudo H, Kikuchi A et al. Genetic heterogeneity in 26 infants with a hypomyelinating leukodystrophy. Hum Genet 2016; 135: 89–98.
Pyle A, Smertenko T, Bargiela D, Griffin H, Duff J, Appleton M et al. Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain 2015; 138: 276–283.
Sagnelli A, Magri S, Farina L, Chiapparini L, Marotta G, Tonduti D et al. Early-onset progressive spastic paraplegia caused by a novel TUBB4A mutation: brain MRI and FDG-PET findings. J Neurol 2016; 263: 591–593.
Carvalho D, Santos S, Martins B, Marques FP . TUBB4A novel mutation reinforces the genotype-phenotype correlation of hypomyelination with atrophy of the basal ganglia and cerebellum. Brain 2015; 138: e327.
Löwe J, Li H, Downing KH, Nogales E . Refined structure of alpha beta-tubulin at 3.5 A resolution. J Mol Biol 2001; 313: 1045–1057.
Blumkin L, Halevy A, Ben-Ami-Raichman D, Dahari D, Haviv A, Sarit C et al. Expansion of the spectrum of TUBB4A-related disorders: a new phenotype associated with a novel mutation in the TUBB4A gene. Neurogenetics 2014; 15: 107–113.
Vanderver A, Simons C, Helman G, Crawford J, Wolf NI, Bernard G et al. Whole exome sequencing in patients with white matter abnormalities. Ann Neurol 2016; 79: 1031–1037.
Data Citations
Yamamoto, Toshiyuki HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.1396 (2017)
Acknowledgements
We would like to express our gratitude to the patient and his family for their cooperation. This work was mainly supported by the Practical Research Project for Rare/Intractable Diseases from Japan Agency for Medical Research and Development (AMED) and Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number 15K09631 (TY). This work was also supported in part by Japan China Sasakawa Medical Fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Human Genome Variation website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Lu, Y., Ondo, Y., Shimojima, K. et al. A novel TUBB4A mutation G96R identified in a patient with hypomyelinating leukodystrophy onset beyond adolescence. Hum Genome Var 4, 17035 (2017). https://doi.org/10.1038/hgv.2017.35
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2017.35
This article is cited by
-
In silico analysis of TUBA4A mutations in Amyotrophic Lateral Sclerosis to define mechanisms of microtubule disintegration
Scientific Reports (2023)
-
A TUBB4A Met363Thr variant in pediatric hypomyelination without atrophy of the basal ganglia
Human Genome Variation (2022)
-
In-silico phenotype prediction by normal mode variant analysis in TUBB4A-related disease
Scientific Reports (2022)
-
Independent occurrence of de novo HSPD1 and HIP1 variants in brothers with different neurological disorders – leukodystrophy and autism
Human Genome Variation (2018)
