
Abstract
Orofaciodigital syndrome type 1 or oral–facial–digital syndrome type 1 (OFDS1, OMIM #311200) is an X-linked malformation syndrome caused by hemizygous mutations in the OFD1 (OMIM #300170) gene with presumed male lethality. Recently males with OFDS1 and mutations in OFD1 have been described. We report a 17-year-old male with molar tooth sign, small cerebellum with absence of the cerebellar vermis, complex polydactyly with a Y-shaped metacarpal, renal failure and craniofacial anomalies caused by a novel splice-mutation (c.1129+4A>T) in the OFD1 gene identified by exome sequencing.
Similar content being viewed by others


OFDS1 is a severe X-linked dominant malformation syndrome believed to be lethal in males. Severely affected heterozygous females have been described. It is characterized by polycystic kidney changes, oral and facial anomalies including oral hamartomas and frenulae, cleft palate, malformations of the brain, such as molar tooth sign, agenesis of corpus callosum, cerebellar anomalies, dysmorphic features and various forms of polydactyly.
Recently, males with mutations in OFD1 have been reported. Tsurusaki et al.1 used exome sequencing to detect a novel splice site mutation (c.2388+1G>C) in both a mother and her male neonate.1 Their family history was significant for an unclassified X-linked lethal congenital malformation syndrome with facial, oral and digital malformations common in (oral–facial–digital syndrome type 1) oral-facial-digital syndrome type 1 (OFDS1). In addition, congenital heart defects, genitourinary malformations, situs inversus and ophthalmological abnormalities were noted. All affected males died within the first 14 days of life. Thauvin-Robinet2 performed sequencing of OFD1 and identified two novel truncating mutations in males whose clinical phenotype was clinically described as an overlap between Joubert syndrome and Simpson–Golabi–Behmel syndrome type 2 (SGBS2, OMIM #312870).2 Both patients had macrocephaly, postaxial polydactyly and a molar tooth sign on cranial magnetic resonance imaging.
We report a 17-year-old male with intellectual disability, seizure disorder, polydactyly, brain malformations, renal failure, small optic nerves with colobomas, and oral and facial anomalies. This child was the product of a term gestation to a 32-year-old healthy mother and a 33-year-old healthy father. This couple previously terminated a female twin gestation pregnancy due to multiple anomalies for which no autopsy or testing was performed. Two maternal half-brothers, now 24 and 32-year-old, were healthy. Both parents have an otherwise negative family history with respect to congenital anomalies, recurrent miscarriages or known genetic conditions. There was no consanguinity; the parents are of Hispanic descent.
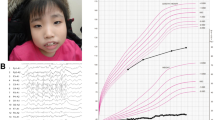
The pregnancy was uneventful with normal prenatal screening and ultrasounds. Birth weight was 3,359 gm (25–50th centile), birth length 43.5 cm (<3rd centile) and birth occipitofrontal circumference 37.5 cm (75–90th centile). APGAR scores were 2 and 8 at 1 and 5 min, respectively, requiring immediate intubation. Physical examination after birth identified oral and tongue hamartomas (Figure 1), pre-auricular pits and cleft palate. Examination of the limbs identified mesoaxial polydactyly of hands and preaxial polydactyly of feet bilaterally (Figure 1). He developed intractable seizures on day of life three. While in the newborn intensive care unit, he was diagnosed with epiglottic agenesis requiring tracheostomy and gastrostomy tube placement at 3 weeks of age. Intestinal malrotation was discovered during preoperative evaluation for G-tube placement and repaired at 3 weeks of age. He failed his newborn hearing screen and brainstem auditory evoked response demonstrated profound sensorineural hearing loss. Ophthalmologic evaluation showed small optic nerves, optic nerve colobomas and absent visual evoked potentials. Hypothyroidism was diagnosed and treated at 18 months of age.

(a) Hamartoma of the tongue, repaired cleft palate and bifrontal narrowing in the patient with c.1129+4A>T OFD1 variant. (b) Preaxial polydactyly with bifid halluces. (c) Magnetic resonance imaging shows a small cerebellum and vermis with a Dandy Walker malformation with enlarged posterior fossa. The patient also had ventriculomegaly and absence of the corpus callosum. (d) Molar tooth sign.
On physical examination at 3 years and 7 months old, his length was 85.5 cm (<3rd centile for age and at the 50th centile for 21 months of age), weight was 21.6 kg (>95th centile for age and at the 50th centile for 5 and 1/2 years of age), occipitofrontal circumference was 50.5 cm (50th centile for age). The patient was non-verbal and significantly developmentally delayed with no head control or ability to sit independently. Head was symmetric with bifrontal narrowing. Disconjugate movement of the eyes and nystagmus were noted. Examination of the mouth was remarkable for hamartomas of the tongue and repaired cleft palate. Echocardiography was normal. Cranial magnetic resonance imaging demonstrated molar tooth sign, small cerebellum and vermis and Dandy Walker malformation with enlarged posterior fossa, marked ventriculomegaly and absence of the corpus callosum (Figure 1). By the age of 5 years, he had developed renal failure with nephrocalcinosis on ultrasound and dialysis dependency. He was dependent on a tracheal stoma on room air, and a gastrostomy tube for full feeding. He had intractable seizures and apparent intellectual disability at 17 years.
Previous genetic testing included a normal karyotype. A microarray was not performed. Sanger sequencing for GLI3 (OMIM #165240) was performed based on overlapping features with phenotypes caused by GLI3 including Pallister–Hall syndrome (OMIM #146510) and Greig Cephalopolysyndactyly syndrome (OMIM #175700). No pathogenic variant in the GLI3 gene was identified.
As a part of an effort to expand the mutational spectrum of patients with overlapping features of oral–facial–digital phenotypes associated with mutations in GLI3, exome sequencing was performed on this patient and his parents as described.3 This study was reviewed and approved by the National Human Genome Research Institute (NHGRI) Institutional Review Board. Informed consent was obtained from the family.
Variants were initially filtered for predicted loss of function including frameshift, nonsense and splice site alterations. The resulting set of variants was further filtered for absence from 938 controls. A novel splice site mutation in exon 11 of the OFD1 gene, NM_003611.2: c.1129+4A>T, was identified in the proband and his mother (Figure 2). Three variant reads were identified in the mother out of a total read depth of 18 suggesting 17% mosaicism. Sanger sequence confirmation was performed as described with a reduced signal from the mutant allele supporting mosaicism.4 This novel splice site variant was not present in the Human Gene Mutation Database (HGMD Version 2014.2) or in the Exome Variant Server (Version ESP6500SI-V2, 6,503 individuals, Seattle, WA, USA; http://evs.gs.washington.edu/EVS). In addition, it was not present in 938 internal control individuals (ClinSeq data set, Bethesda, MD, USA).

Electropherogram of c.1129+4A>T identified in the patient and his mother (mosaic).
The c.1129+4A>T variant was scored as deleterious by two splice predictor algorithms, Human Splicing Finder and MaxEntScan. Reverse transcription polymerase chain reaction (OneStep, Qiagen, Germantown, MD, USA) analysis of total RNA isolated from patient lymphoblasts (RNeasy, Qiagen) identified the normal splice product in addition to two alternate splice products resulting from exon skipping of either exon 11 or exons 11 and 12 (Figure 3). Both alternative splice products are predicted to result in frameshifts. Splicing out exon 11 predicts p.Tyr353Lysfs*13 and splicing out exons 11 and 12 predicts p.Lys352Asnfs*4 (NM_003611.2 reference transcript).

Agarose gel of reverse transcription polymerase chain reaction products from lymphoblast RNA from patient (M33; c.1129+4A>T) and a control. Wild-type splicing was evident in both samples. In addition, two alternate splice products were identified in the patient resulting in exon skipping of exon 11 (green), or exon 11 and 12 (orange) as diagramed. Products were confirmed by Sanger sequencing.
Each product was isolated by gel extraction (QIAquick, Qiagen) and directly sequenced in both directions on a 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). It is likely that the non-lethal phenotype seen in the proband, and potentially the lack of phenotype in the mother, is due to the presence of the normal splice product. In addition, X-inactivation studies were performed using both methylation sensitive polymerase chain reaction with bisulfite modification of genomic DNA (EpiTect, Qiagen) and the human androgen receptor (HUMARA) assay as described with minor modifications.5,6 Both assays showed >94% skewing in the mother (data not shown) potentially contributing to her lack of phenotype.
In summary, we report a novel variant in OFD1 in a 17-year-old male and his unaffected mother supporting recently published reports documenting surviving affected males with OFDS1. The mother was mosaic for the mutation and did not have dysmorphic features or oral findings. This proband was initially diagnosed with OFDS type VI also known as Varadi–Papp syndrome, an autosomal recessive condition characterized by small or absent cerebellar vermis, oral hamartomas, facial dysmorphism and mesoaxial polydactyly. The gene most commonly mutated in patients with this condition is C5orf42 (OMIM #614571). Other genes reported with this condition are OFD1 and TMEM216 (OMIM #613277). The patient reported here has these features and in addition malrotation, hearing loss, small optic nerves and renal failure with nephrocalcinosis based on ultrasound examination. It very likely his nephronophtisis based on his ciliary dysplasia. We suspect the malrotation could also be associated with the described situs inversus in a patient previously reported by other authors.2 A newer classification of Joubert syndrome and related disorders with the molar tooth sign suggested Varadi–Papp syndrome should be redesignated as Joubert syndrome with oro–facio–digital defects.7 This subtype of Joubert syndrome comprises typical Joubert syndrome features with tongue anomalies (bifid or lobulated tongue), oral hamartomas or frenulae, and forms of polydactyly, most often mesoaxial polydactyly.
Over time, the phenotypic parameters of recognizable disorders have slowly expanded as clinicians recognize progressively less typically affected patients, thereby enlarging the clinical spectrum of the disease.
Next-generation sequencing approaches now allow and massively accelerate the process of determining a clinical diagnosis because essentially all genes are tested at once, often more cost-effective, rather than a sequential testing process of individual genes. This allows us to more rapidly recognize atypical patients and better identify patients with highly atypical presentations. As in the patient described here, it also allows us to identify the correct diagnosis in patients with phenotypes with overlapping features or overlapping clinical definitions and classifications. Above all, this teaches us an increasing appreciation of the phenotypic spectrum of human genomic variation and a much greater pleiotropic variety of individual conditions as shown in this example, a male patient with OFDS1 who was classified as OFDS type VI for years based on clinical findings.
Until recently, OFDS1 was presumed to be lethal in males with a severe phenotype in heterozygous females. We recognize that affected males with OFDS1 do in fact survive in some cases. Our report along with recently published observations expands the previous understanding of the phenotypic spectrum of this disorder. We hypothesize that the clinical spectrum of the disorder and range of mutations is wider than that represented by the patients identified to date, especially in male patients and their mildly affected, atypically affected or unaffected carrier mothers. We speculate that the splice variant identified in this patient produces a mix of mutant and wild-type transcript, and that the latter is responsible for the non-lethal phenotype. Finally, we add to the mutational spectrum of the disorder with this novel variant in the OFD1 gene. The use of next-generation sequencing will aid in expanding the phenotypic spectrum of disorders like OFDS1 as more patients with non-specific or atypical clinical features are analyzed and correctly diagnosed. It furthermore improves our understanding of the biology of allelic disorders with overlapping features or overlapping definitions and disease classifications.
We look forward to future ascertainment of additional patients with this condition to lead to a more complete understanding of the clinical spectrum, facilitate its clinical recognition and appreciate the remarkable developmental pleiotropy of OFD1 dysfunction and phenotype.
References
References
Tsurusaki Y, Kosho T, Hatasaki K, Narumi Y, Wakui K, Fukushima Y et al. Exome sequencing in a family with an X-linked lethal malformation syndrome: clinical consequences of hemizygous truncating OFD1 mutations in male patients. Clin Genet 2013; 83: 135–144.
Thauvin-Robinet C, Thomas S, Sinico M, Aral B, Burglen L, Gigot N et al. OFD1 mutations in males: phenotypic spectrum and ciliary basal body docking impairment. Clin Genet 2013; 84: 86–90.
Johnston JJ, Sapp JC, Curry C, Horton M, Leon E, Cusmano-Ozog K et al. Expansion of the TARP syndrome phenotype associated with de novo mutations and mosaicism. Am J Med Genet 2014; 164A: 120–128.
Gripp KW, Hopkins E, Johnston JJ, Krause C, Dobyns WB, Biesecker LG . Long-term survival in TARP syndrome and confirmation of RBM10 as the disease-causing gene. Am J Med Genet 2011; 155A: 2516–2520.
Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW . Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 1992; 51: 1229–1239.
Kubota T, Nonoyama S, Tonoki H, Musuno M, Imaizumi K, Kojima M et al. A new assay for the analysis of X-chromosome inactivation based on methylation-specific PCR. Hum Genet 1999; 104: 49–55.
Brancati F, Dallapiccola B, Valente EM . Joubert Syndrome and related disorders. Orphanet J Rare Dis 2010; 5: 20.
Data Citations
Wentzensen, Ingrid HGV Database (2015) http://dx.doi.org/10.6084/m9.figshare.hgv.768
Acknowledgements
We thank the families for their participation in this study. This work was supported by funding from the Intramural Research Program of the National Human Genome Research Institute of the US National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Wentzensen, I., Johnston, J., Patton, J. et al. Exome sequencing identifies a mutation in OFD1 in a male with Joubert syndrome, orofaciodigital spectrum anomalies and complex polydactyly. Hum Genome Var 3, 15069 (2016). https://doi.org/10.1038/hgv.2015.69
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.69
This article is cited by
-
Clinical spectrum of male patients with OFD1 mutations
Journal of Human Genetics (2019)
