
Abstract
Severe congenital protein C (PC) deficiency is an autosomal recessive hereditary thrombophilia caused by mutations in PROC. The case manifested severe purpura fulminans, intracranial thrombosis or hemorrhage within 4 days after birth, resulting in blindness. We report the identification of inherited compound heterozygous mutations, including a novel nonsense mutation in PROC, and a prenatal genetic test for a subsequent pregnancy. Prenatal diagnosis may facilitate preemptive and radical therapy for severe PC deficiency.
Similar content being viewed by others


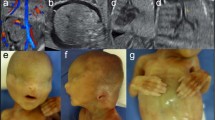
Protein C (PC) is the proenzyme of a vitamin K-dependent serine protease involved in blood coagulation. Deficiency in PC impairs the inactivation of activated factors V and VIIIC, promoting excessive fibrin formation and constituting a risk factor for thrombosis.1 Severe congenital PC deficiency (OMIM: 612304) is rare, although heterozygous PC deficiency (OMIM: 176860) is the second leading cause of genetic predisposition to thrombosis in Japan.2 FV Leiden (G1691A) and FII mutation (G20210A) are the major causes of thrombosis in Caucasians.3,4 However, in Asian populations, approximately half of the patients with deep venous thrombosis carried heterozygous mutations in one of the protein S (PS; PROS1), PC (PROC) or antithrombin (AT; SERPINC1) genes.5–7 Mutations in PROS1, PROC and SERPINC1 have larger effects on thrombosis than mutations in FV Leiden or FII.8 Although it is still controversial whether thrombosis due to these genetic conditions is a risk factor for pregnancy complications, the risk for thrombosis during pregnancy is high.9 The frequency of heterozygous mutations in PROC in the Japanese population has previously been estimated at 1 in 700 based on the plasma activity of PC in a selected population.10 None of the parents of patients with congenital PC deficiency reported in this study had experienced thromboembolism.10 However, considering the frequency of congenital PC deficiency in Japan, the true frequency of heterozygous mutations may be higher.5 Here we report a familial case of PC deficiency with a novel mutation in PROC, and a prenatal genetic test for severe congenital PC deficiency.
A genetic study was conducted for the family. Informed consent was obtained from the proband’s parents. Experimental protocols and ethics approval for this study was obtained from the Review Boards of Kobe University. Peripheral blood samples were obtained from the family. Genomic DNA was extracted using the QIAamp DNA Mini Kit (Qiagen, San Francisco, CA, USA). Targeted Sanger sequencing was performed to detect mutations in the PROC gene.10 Primers for sequencing were PROC_exon9_F: 5′-AGTGCCTGGGACGTGTGGGT-3′; PROC_exon9_R: 5′-GTGCCGTGGAAGGAGGCGAC-3′. For prenatal diagnosis, amniotic fluid was collected at 16 weeks of gestation. DNA was directly extracted from 10 ml of amniotic fluid. To exclude contamination of the maternal genome, six microsatellite markers were analyzed for the family.11
The index case was a 3-year-old boy with total blindness. No intellectual disability or psychomotor developmental delay was demonstrated. The proband, the first child of nonconsanguineous parents, was born at 38 weeks and 4 days of gestation. His father had bilateral amblyopia and right hydronephrosis at the age of 10. His mother had no past medical history until she had a massive hemorrhage during delivery, for which she needed a blood transfusion. Both great-grandfathers died from cerebral infarction (Figure 1b). The patient was transferred to the newborn intensive care unit at Kobe University Hospital 2 days post birth presenting with dark purpura on both sides of his legs (Figure 1a, upper left and middle). Well-demarcated purpuric lesions with central necrosis suggested a diagnosis of purpura fulminans (PF). Bilateral ocular hemorrhage and intracranial thrombosis or hemorrhage (Figure 1a, upper and lower right) were observed on day 3. Blood tests showed that PC activity and antigen were below 10 and 5%, respectively. Hemoglobin level was 11.2 g dl−1; white blood cell count was 11,400 μ l−1; and platelet count was 70,000 μ l−1. Prothrombin time per international normalized ratio (1.41) and activated partial thromboplastin time (46.5 s) were slightly elevated. Plasma fibrinogen level was decreased (41.0 mg dl−1) but D-dimer was increased (111.1 μg ml−1), as were plasma fibrinogen degradation products (203.2 μg ml−1). Both antithrombin level (40%, normal range 60–90%) and PS free antigen level (55%, normal range 60–150%) were slightly decreased. Therefore, PF as a result of congenital severe PC deficiency with disseminated intravascular coagulation was suspected. The patient was started on anticoagulation therapy on day 3: 30 ml kg−1 of fresh frozen plasma (FFP) was transfused. PC (12,500 units) (Anact C, The Chemo-Sero-Therapeutic Research Institute, Japan) was also transfused continuously from day 4 to 9. From day 10, 0.1 mg kg day−1 of warfarin (Eisai, Tokyo, Japan) was administered, with gradual increase of the dosage. Symptoms were markedly improved, with PF completely resolved by day 14 (Figure 1a, lower left and middle). Target sequencing for PROC identified compound heterozygous mutations c.1015G>A (p.Val339Met) and c.1003C>T (p.Gln335*) in exon 9 of PROC (Figure 1b). The mutations were detected in the mother and father, respectively. The c.1015G>A mutation detected in the mother was previously reported as a founder mutation;12 however, the c.1003C>T mutation was novel. Blood tests for the parents showed that PC activity and antigen were decreased in both: 53 (normal range 64–146%) and 35% (normal range 70–150%) in the father and 50 and 43% in the mother, respectively.

(a) Images of the patient taken on day 2 (upper) and day 10 (lower) post birth. Photograph showing demarcation of healthy and ischemic skin. Necrosis extended to the tendons (arrow) of right lower leg because of purpura fulminans (PF). PF was completely resolved after anticoagulation therapy (lower left and middle). Computed tomography (CT) image (day 2) and magnetic resonance imaging (MRI, day 3) of brain, coronal view, showing extensive intracranial thrombosis or hemorrhage in the right occipital lobe. Right upper panel: CT imaging of the brain. Right lower panel: MRI. (b) Familial pedigree. Squares: males; circles: females; open shape: unaffected, noncarrier; half-filled shape: heterozygous carrier; filled shape: congenital protein C deficiency patient. Both parents carried heterozygous mutations in PROC. (c) Prenatal genetic analysis for the fetus. The fetus inherited a wild-type allele from the father and a missense mutation from the mother.
The parents consulted the genetic counseling unit at the Kobe University Hospital about the possibility of prenatal genetic testing in their next pregnancy. Board members discussed the ethics of the test and determined that they would offer the prenatal test, but only provide the results of the diagnosis if the fetus was found to be a compound heterozygote. After genetic counseling, prenatal testing was performed. The fetus was a heterozygous carrier of the maternal pathogenic allele (Figure 1c). We informed the parents that the fetus was not a compound heterozygote of the mutations and therefore would not have the symptoms of severe PC deficiency, and the parents continued the pregnancy. During the pregnancy, the mother’s PC activity and antigen level decreased to between 29–51% and 33–55%, respectively. Usually, the plasma activity levels of PS decrease but those of PC increase until 22 weeks of gestational age.13 However, PC activity levels remained low throughout the pregnancy. The fetus was delivered at 38 weeks of gestation by an uncomplicated vaginal delivery. The mother was prescribed warfarin after the pregnancy. The baby was female, with birth weight 2,646 g. Although her PC activity and antigen level was also decreased to 16 and 17%, respectively, she did not show any signs of thrombosis, and was clinically healthy. The genetic diagnosis for the fetus was confirmed phenotypically and genetically after birth.
Severe congenital PC deficiency is a rare genetic condition with severe thrombosis, manifesting systemic symptoms, sometimes soon after birth, with poor prognosis, even when the patient is treated with FFP, anticoagulants and antibody-purified PC concentrate.2 In contrast, a milder, autosomal dominant, PC deficiency is frequently seen in Asian populations. Surprisingly, the incidence of venous thromboembolism for heterozygous carriers of PROC mutations in Asia was estimated at 40%.14 Despite the high prevalence of PC deficiency, maternal screening for coagulation factors is not common in Asia. Moreover, there are few parents with PC deficiency who manifest thrombosis until reproductive age. Therefore, it is quite difficult to predict severe congenital PC deficiency before birth. Indeed, several cases of prenatal diagnosis for congenital PC deficiency have been reported worldwide, but for subsequent pregnancies.15,16
This is the first case of prenatal diagnosis of PC deficiency reported in Japan. Recently, domino liver transplantation for the treatment of congenital PC deficiency has been reported.17 New interventions such as this, or regenerative medicines, will be better implemented if treatment is initiated in a timely manner owing to earlier diagnosis with prenatal genetic testing.
More than 200 mutations affecting PC have been reported worldwide.18 In the present case, both mutations were in the active site of PROC.12 The p.Val339Met mutation found in the mother is one of the main founder mutations in the Japanese population,19 but it is not found in the Korean population.14 This implies that this mutation arose in recent years, given that Japanese and Korean populations originate from the same ancestors. Considering the high carrier frequency of this mutation in the Japanese population, its expansion over such a short period of time may mean that some advantage is conferred on carriers of this mutation. Although the father’s mutation was a novel nonsense mutation, his previous medical history and serum PC level indicate that this mutation is likely pathogenic. Offering a precise questionnaire for familial medical history may have provided clues for the identification of PC deficiency in the patient.
In conclusion, we have identified a novel missense mutation in PROC, which leads to severe PC deficiency. Despite timely diagnosis and treatment, the case did not escape the complication of blindness. Genetic testing was able to provide the family with the possibility of family planning, or earlier treatment options. In the event of a pathogenic diagnosis, more radical treatment for congenital PC deficiency could be expected.
References
References
Broekmans AW, Veltkamp JJ, Bertina RM . Congenital protein C deficiency and venous thromboembolism. A study of three Dutch families. N Engl J Med 1983; 309: 340–344.
Ohga S, Kang D, Kinjo T, Ochiai M, Doi T, Ishimura M et al. Paediatric presentation and outcome of congenital protein C deficiency in Japan. Haemophilia 2013; 19: 378–384.
Irani-Hakime N, Tamim H, Kreidy R, Almawi WY . The prevalence of factor V R506Q mutation-Leiden among apparently healthy Lebanese. Am J Hematol 2000; 65: 45–49.
Rosendaal FR, Doggen CJ, Zivelin A, Arruda VR, Aiach M, Siscovick DS et al. Geographic distribution of the 20210 G to A prothrombin variant. Thromb Haemost 1998; 79: 706–708.
Ohga S, Ishiguro A, Takahashi Y, Shima M, Taki M, Kaneko M et al. Protein C deficiency as the major cause of thrombophilias in childhood. Pediatr Int 2013; 55: 267–271.
Kinoshita S, Iida H, Inoue S, Watanabe K, Kurihara M, Wada Y et al. Protein S and protein C gene mutations in Japanese deep vein thrombosis patients. Clin Biochem 2005; 38: 908–915.
Miyata T, Sato Y, Ishikawa J, Okada H, Takeshita S, Sakata T et al. Prevalence of genetic mutations in protein S, protein C and antithrombin genes in Japanese patients with deep vein thrombosis. Thromb Res 2009; 124: 14–18.
Neki R, Miyata T, Fujita T, Kokame K, Fujita D, Isaka S et al. Nonsynonymous mutations in three anticoagulant genes in Japanese patients with adverse pregnancy outcomes. Thromb Res 2014; 133: 914–918.
Sakata T, Kario K, Katayama Y, Matsuyama T, Kato H, Miyata T . Studies on congenital protein C deficiency in Japanese: prevalence, genetic analysis, and relevance to the onset of arterial occlusive diseases. Semin Thromb Hemost 2000; 26: 11–16.
Sakata T, Okamoto A, Mannami T, Matsuo H, Miyata T . Protein C and antithrombin deficiency are important risk factors for deep vein thrombosis in Japanese. J Thromb Haemost 2004; 2: 528–530.
Kondo-Iida E, Kobayashi K, Watanabe M, Sasaki J, Kumagai T, Koide H et al. Novel mutations and genotype-phenotype relationships in 107 families with Fukuyama-type congenital muscular dystrophy (FCMD). Hum Mol Genet 1999; 8: 2303–2309.
Gandrille S, Aiach M . Identification of mutations in 90 of 121 consecutive symptomatic French patients with a type I protein C deficiency. The French INSERM Network on molecular abnormalities responsible for protein C and protein S deficiencies. Blood 1995; 86: 2598–2605.
Bereczky Z, Kovacs KB, Muszbek L . Protein C and protein S deficiencies: similarities and differences between two brothers playing in the same game. Clin Chem Lab Med 2010; 48 (Suppl 1): S53–S66.
Kim HJ, Seo JY, Lee KO, Bang SH, Lee ST, Ki CS et al. Distinct frequencies and mutation spectrums of genetic thrombophilia in Korea in comparison with other Asian countries both in patients with thromboembolism and in the general population. Haematologica 2014; 99: 561–569.
Millar DS, Allgrove J, Rodeck C, Kakkar VV, Cooper DN . A homozygous deletion/insertion mutation in the protein C (PROC) gene causing neonatal Purpura fulminans: prenatal diagnosis in an at-risk pregnancy. Blood Coagul Fibrinolysis 1994; 5: 647–649.
Alessi MC, Aillaud MF, Paut O, Roquelaure B, Alhenc-Gelas M, Pellissier MC et al. Purpura fulminans in a patient homozygous for a mutation in the protein C gene--prenatal diagnosis in a subsequent pregnancy. Thromb Haemost 1996; 75: 525–526.
Matsunami M, Ishiguro A, Fukuda A, Sasaki K, Uchida H, Shigeta T et al. Successful living domino liver transplantation in a child with protein C deficiency. Pediatr Transplant 2015; 19: E70–E74.
D'Ursi P, Marino F, Caprera A, Milanesi L, Faioni EM, Rovida E . ProCMD: a database and 3D web resource for protein C mutants. BMC Bioinformatics 2007; 8 (Suppl 1): S11.
Miyata T, Sakata T, Yasumuro Y, Okamura T, Katsumi A, Saito H et al. Genetic analysis of protein C deficiency in nineteen Japanese families: five recurrent defects can explain half of the deficiencies. Thromb Res 1998; 92: 181–187.
Data Citations
Taniguchi-Ikeda, Mariko HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.588 (2015)
Taniguchi-Ikeda, Mariko HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.590 (2015)
Acknowledgements
We greatly appreciate the patients’ and their family’s cooperation.
Author information
Authors and Affiliations
Contributions
IM, AS, TM, IK and AH took care of the proband and wrote the manuscript. MTI and ST took care of the whole family for genetic counseling, prenatal diagnosis, performed experiments and wrote the manuscript. YO, YN, YN, SK and TE performed genetic analysis for the whole family and wrote the manuscript. HY, TT, HK and KI attended the planning of the project and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Tairaku, S., Taniguchi-Ikeda, M., Okazaki, Y. et al. Prenatal genetic testing for familial severe congenital protein C deficiency. Hum Genome Var 2, 15017 (2015). https://doi.org/10.1038/hgv.2015.17
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.17
This article is cited by
-
Cascade health service use in family members following genetic testing in children: a scoping literature review
European Journal of Human Genetics (2021)
