
Abstract
We report a large, non-consanguineous family comprising five generations of individuals residing in Gujarat, India affected with localized Epidermolysis Bullosa Simplex (EBS) Koebner type. We analyzed 14 individuals including 9 affected individuals from this family. Exome sequencing in two cases suggested a novel non-synonymous variation, p.L325H, in the KRT5 gene. The present analysis also reports the first causative mutation of EBS Koebner type from India.
Similar content being viewed by others


Report
Epidermolysis bullosa simplex (EBS) is a rare and polymorphic skin disorder characterized by skin fragility, which causes blisters and further erosion owing to mechanical pressure or friction. The pathophysiology of this disorder has been traced to the fragility of the skin layer organization, primarily attributed to abnormal structure and/or architecture of keratin microfilaments in the skin, resulting in skin that is fragile and sensitive to mechanical pressure or friction. This precipitates separation of the epidermis and dermis, which leads to the formation of bullae.1,2 The disorder can manifest as a wide spectrum of polymorphic phenotypes with varying severity. A number of distinct clinical subtypes of the disease have been reported that range from localized phenomena, which was previously known as the Weber–Cockayne variant where the blisters are usually limited to the hands, soles and other pressure points, to generalized bullous disorder, otherwise known as the Dowling–Meara variant and the Koebner variant. Phenotypic variants of the disorder may also co-occur with pigmentation problems, also referred to as the mottled pigmentation variant. EBS is a rather rare genetic disorder with prevalence estimates of ~1 in 25,000–50,000 individuals. The inheritance of the disease generally follows an autosomal dominant pattern, although in a minor set of mutations, an autosomal recessive pattern has been observed.3
A number of mutations have been characterized in the Keratin 5 (KRT5) and Keratin 14 (KRT14) genes, which form a heterodimeric complex constituting the keratin cytoskeleton that forms the basal cells of the skin. Recently, additional mutations in the plectin gene (PLEC1) encoding the protein plectin have also been implicated as one of the causative markers for EBS4. The mutation frequency in KRT5 and KRT14 distinctly vary in each of the EBS clinical subtypes, with localized EBS caused predominantly by variations in the KRT5 gene and the autosomal recessive form of the disease caused predominantly by variants in the KRT14 gene.3,5
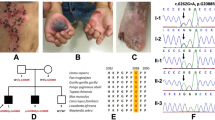
In the present study, we analyzed a large, non-consanguineous five-generation family residing in Gujarat, India (Figure 1d). Blood samples were obtained, after informed consent, from a total of 14 individuals, out of which 9 were affected and 5 were apparently unaffected. The affected individuals had classical EBS features, with the bullae confined primarily to the palms, soles and pressure points including the waistline (Figures 1a–c). Almost all individuals had hyperkeratosis and patches of hyperpigmentation on the limb extremities, but no family members had reticulate hyperpigmentation in any of the flexural regions. The nails were also observed to be affected in a number of individuals, and the affected members did not have large phenotypic variability.

Identification of a novel mutation associated with EBS in a large Gujarati family. (a–c) Clinical features of the affected individuals with bullae and erosion. (d) Pedigree of the family. (e) Chromatogram derived from targeted capillary sequencing of 14 samples for mutation p.L325H. Affected individuals are marked with asterisks. (f) Schematic description of KRT5 gene showing mutation loci, secondary structure with domain annotations.
Whole-exome capture sequencing was performed with 101 bp-paired end reads for two affected individuals in the family using Illumina exome capture and sequencing on an Illumina HiSeq 2000 platform (San Diego, CA, USA). We generated over 82 and 63 million reads each for the two affected individuals (SRA accession SRR1380978 (III:4) and SRR1380977 (IV:4)). Variants were independently called using standard mapping and variant calling protocols, which involved bwa6 for mapping reads and samtools7 for calling variants. The variants were further annotated using ANNOVAR,8 analyzed via SIFT for potentially deleterious non-synonymous variations by using the SIFT score (<0.05)9 and the presence of the variant was verified in both affected individuals. The analysis revealed a total of three putative deleterious variants in the laminin gamma 2 (LAMC2), collagen type XVII (COL17A1) and KRT5 genes. The non-synonymous variation p.L325H (c.T974A) in the KRT5 gene was heterozygous in both affected individuals. Further, the region flanking the variant was genotyped in all the 14 samples by capillary sequencing using BigDye-terminator chemistry (Figure 1e). Only the non-synonymous variation p.L325H segregated in the affected individuals, suggesting high penetrance.
In addition, we performed an in-depth in silico analysis of this particular variant. Our analysis revealed that the p.L325H mutation is located in a highly conserved region of the KRT5 gene. The keratin protein has distinct structural and functional domains that have differential mutation rates. In brief, the entire protein comprises six distinct structural and functional motifs. These motifs are the head, helix initiation motif, coils, linkers, helix termination motif and tail (Figure 1f). In this particular case, secondary structure analysis revealed that the mutation occurs in the Linker-12 region of the protein. A number of deleterious mutations have been mapped to the KRT5 gene and have been previously associated with the EBS phenotype in the Linker-12 region. A number of variants also have different penetrance and different modes of inheritance, but the majority of mutations have an autosomal dominant inheritance pattern.10,11 A mutation in the same locus with an A>C substitution has been previously reported and is associated with the disease phenotype.12
In the present analysis, we identified a novel variant, p.L325H (c.T974A), in the KRT5 gene that is associated with EBS in a large family, comprising five generations and over 57 individuals. In addition to advancing the mutation spectrum of the EBS phenotype, we demonstrated that this novel variant may disrupt the L-12 domain of the KRT5 gene. The present study also details the largest family with EBS reported from India.
References
References
Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A et al. The classification of inherited Epidermolysis Bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol 2008; 58: 931–950.
Coulombe PA, Kerns ML, Fuchs E . Epidermolysis bullosa simplex: a paradigm for disorders of tissue fragility. J Clin Invest 2009; 119: 1784–1793.
Szeverenyi I, Cassidy AJ, Chung CW, Lee BT, Common JE, Ogg SC et al. The Human Intermediate Filament Database: comprehensive information on a gene family involved in many human diseases. Hum Mutat 2008; 29: 351–360.
Koss-Harnes D, Høyheim B, Anton-Lamprecht I, Gjesti A, Jørgensen RS, Jahnsen FL et al. A site-specific plectin mutation causes dominant epidermolysis bullosa simplex Ogna: two identical de novo mutations. J Invest Dermatol 2002; 118: 87–93.
Yasukawa K, Sawamura D, McMillan JR, Nakamura H, Shimizu H . Dominant and recessive compound heterozygous mutations in epidermolysis bullosa simplex demonstrate the role of the stutter region in keratin intermediate filament assembly. J Biol Chem 2002; 277: 23670–23674.
Li H, Durbin R . Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2009; 25: 1754–1760.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
Wang K, Li M, Hakonarson H . ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data. Nucl Acids Res 2010; 38: e164.
Kumar P, Henikoff S, Ng PC . Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4: 1073–1081.
Goldsmith LA . Mutations in Epidermolysis Bullosa Simplex. J Invest Dermatol 1995; 105: 529–531.
Coulombe PA, Lee CH . Defining keratin protein function in skin epithelia: epidermolysis bullosa simplex and its aftermath. J Invest Dermatol 2012; 132: 763–775.
Sørensen CB, Ladekjaer-Mikkelsen AS, Andresen BS, Brandrup F, Veien NK, Buus SK et al. Identification of novel and known mutations in the genes for keratin 5 and 14 in Danish patients with epidermolysis bullosa simplex: correlation between genotype and phenotype. J Invest Dermatol 1999; 112: 184–190.
Data Citations
Scaria, Vinod HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.493 (2014)
Acknowledgements
The authors acknowledge discussions and help from Ayesha Pasha, Manish Kumar Dwivedi and Sunil Shakya for the preparation of the manuscript and capillary sequencing. This study was funded by the Council of Scientific and Industrial Research, India through Grant No. BSC0122.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Vellarikkal, S., Patowary, A., Singh, M. et al. Exome sequencing reveals a novel mutation, p.L325H, in the KRT5 gene associated with autosomal dominant Epidermolysis Bullosa Simplex Koebner type in a large family from western India. Hum Genome Var 1, 14007 (2014). https://doi.org/10.1038/hgv.2014.7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2014.7
