
Abstract
Iris hypoplasia (IH) is rare autosomal dominant disorder characterized by a poorly developed iris stroma and malformations of the eyes and umbilicus. This disorder is caused by mutation of the paired-like homeodomain 2 (PITX2) gene. Here, we describe a novel PITX2 mutation (c.205C>T) in an IH family presenting with very mild eye features but with tooth agenesis as the most obvious clinical feature.
Similar content being viewed by others


Mutations in the PITX2 (paired-like homeodomain 2) gene are associated with three allelic disorders: iris hypoplasia (IH), iridogoniodysgenesis syndrome (IGDS; OMIM 137600) and Axenfeld-Rieger syndrome (ARS; OMIM 180500).1–3 Various dental abnormalities including tooth agenesis are also found in IH, IGDS and ARS, which are characterized by abnormal development of the anterior segment of the eyes and umbilicus anomalies.1–4 IH shows the mildest phenotype among the three, characterized by only iris hypoplasia and glaucoma.1,5 IGDS presents with goniodysgenesis in addition to iris hypoplasia and glaucoma.2 ARS shows iridocorneal adhesions, a prominent Schwalbe line, and an approximately 50% risk of glaucoma.4
PITX2, also called pituitary homeobox 2, encodes a member of the RIEG/PITX homeobox family, which belongs to the bicoid class of homeodomain proteins. Pitx2 is one of the earliest epithelial markers of tooth development,6 having crucial roles in the genetic control of tooth pattern formation and epithelial differentiation.7–9 Therefore, mutations in PITX2 might affect odontogenesis in IH, IGDS and ARS patients.
In the present study, we used whole-exome sequencing (WES) to identify a novel PITX2 mutation (c.205C>T, p.R69C) in a Japanese family showing tooth agenesis.
Blood samples were obtained from seven individuals from a Japanese family with tooth agenesis. Experimental protocols were approved by the Institutional Review Board of Aichi-Gakuin University, the Institute for Developmental Research, Aichi Human Service Center and Yokohama City University School of Medicine. Informed consent was obtained from all the family members participating in this research.
Genomic DNA was extracted from peripheral blood leukocytes using a PAXgene Blood DNA Kit (PreAnalytiX; Qiagen, Valencia, CA, USA).
Sanger sequencing. The entire coding regions and exon–intron boundaries of MSX1, PAX9, AXIN2 and WNT10A were analyzed as described previously10 except for WNT10A, for which we used original PCR primers (information available on request). PCR products were sequenced using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) with the amplification primers on an ABI PRISM 310 DNA sequencer (Applied Biosystems).
Whole-exome sequencing. WES was performed on three affected individuals (Figure 1a; II-3, III-1 and III-3) and one unaffected individual (II-1) from the family. DNA was captured with a SureSelect Human All Exon V5 Kit (Agilent Technologies, Santa Clara, CA, USA) and sequenced on a HiSeq2000 sequencer (Illumina, San Diego, CA, USA) with 101-bp paired-end reads. Image analysis and base calling were performed by sequence control software real-time analysis and CASAVA software v1.8 (Illumina). Reads were aligned and mapped to the human reference genome sequence (UCSC Genome Browser hg19, GRCh37) using Novoalign 2.08.02 (Novocraft, Selangor, Malaysia; http://www.novocraft.com/main/index.php) and PCR duplicates were removed with Picard tools. Variant calling as well as coverage and depth calculations were performed with the Genome Analysis Toolkit 1.6-5 (http://www.broadinstitute.org/gatk/). The resulting single-nucleotide variants were annotated with ANNOVAR (February 2012) (Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, PA, USA). These variants were filtered based on the autosomal dominant inheritance model. Prediction of the damaging effect of each variant was performed using SIFT, PolyPhen-2 and MutationTaster software as reported previously.11 Sanger sequencing of all candidate variants was performed to confirm familial segregation in all the available family members including I-4, II-2 and III-2 who were not examined by WES.

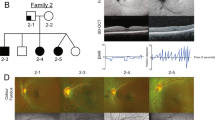
Pedigree, panoramic radiographs and ocular findings. (a) Family pedigree. Squares indicate males and circles indicate females. Black and white symbols indicate affected and unaffected individuals, respectively. The arrow indicates the proband. Asterisks indicate individuals analyzed by whole-exome sequencing. Mut, mutation; wt, wild type. (b) Panoramic radiographs of the proband (III-3). Asterisks indicate tooth agenesis. (c) Ocular findings of proband’s mother (II-3). Arrowheads indicate iris hypoplasia.
Clinical findings. A 17-year-old Japanese girl (III-3) presenting with tooth agenesis was referred to our clinic by her general dental practitioner. Clinical and radiographic dental examination revealed agenesis of 11 permanent teeth (Figure 1b). Furthermore, other family members also showed tooth agenesis. The number of missing permanent teeth was five in her mother (II-3), eight in her brother (III-1) and 12 in her aunt (II-2) (Supplementary Table 1). Information about tooth agenesis in her grandfather, who had severe periodontal disease, could not be obtained. Her mother (II-3) also had maxillary hypoplasia. No additional dental anomalies were noted in any of the affected family members such as microdontia, enamel hypoplasia or craniofacial abnormalities.
Although her grandfather (I-3) was diagnosed as having glaucoma, the proband and her other family members did not complain of any eye-related symptoms. IH, IGDS or ARS were considered retrospectively after obtaining the genetic data. Her mother (II-3) revealed very mild iris stromal hypoplasia and corectopia, as well as moderate glaucoma and severe myopia (Figure 1c), but no iridocorneal adhesions, a prominent Schwalbe line, or goniodysgenesis, supporting IH as the most appropriate diagnosis. No umbilicus anomalies or other health problems were recognized in this family.
Sanger sequencing and WES. No mutations were detected in the four candidate genes (MSX1, PAX9, AXIN2 and WNT10A) for tooth agenesis by Sanger sequencing. By WES of four individuals (II-1, II-3, III-1 and III-3), 1,489 sequence alterations were identified. First, we selected variants shared among all affected individuals and excluded variants observed in the unaffected individual. Second, we excluded synonymous variants or variants found in the dbSNP137 database or our in-house database (n=575). After these two steps, 23 variants remained. We then performed Sanger sequencing of these 23 candidate variants in three other individuals (I-4, II-2 and III-2), after which two candidate variants remained, in PITX2 and CCDC34 (coiled-coil domain containing 34). Both mutations were predicted to be probably disease-causing. The PITX2 mutation (c.205C>T, p.R69C) was more likely to be pathogenic, because PITX2 mutations cause ARS, IGDS and IH, which are associated with tooth agenesis1–3 (Figure 2a). The mutated Arg69 residue is evolutionarily conserved from fish to humans (Figure 2b). Furthermore, Arg69 resides in a highly conserved homeodomain that interacts with DNA (Figure 2c). In addition, three PITX2 transcriptional isoforms are known that differ at the N terminus: A, B and C.3,12–14 This mutation is located in the conserved region among three. The CCDC34 variant is currently of unknown significance as the function of this gene is not well characterized.

Paired-like homeodomain 2 (PITX2) mutation in the family with iris hypoplasia (IH). (a) Electropherograms of the PITX2 mutation. (b) Cross-species multiple alignment of PITX2 protein sequences around the mutation site showing evolutionary conservation of the altered amino acid 69. (c) Schematic presentation of the PITX2 protein with its functional domains and the mutation. H1, helix 1 (residues 48–60); H2, helix 2 (residues 66–75); and H3, helix 3 (residues 80–96). OAR, otp, aristaless and rax-homology domain.
We identified a novel PITX2 mutation c.205C>T (p.R69C) in a Japanese family showing tooth agenesis that was associated with mild ocular symptoms in only one of the affected family members. PITX2 has a crucial role in the formation of eyes, heart and the craniofacial region.3,7 The homeodomain contains three α helices: α1 (residues 48–60), α2 (residues 66–75) and α3 (residues 80–96).15 The R69C mutation is localized in the second helix of the homeodomain (Figure 2c) and the Arg69 residue is one of the contact points to the DNA phosphate backbone.16
Previously, an R69H mutation (the same position as R69C described here) was found in a family with IGDS.2,17 This R69H mutation slightly impaired DNA-binding capacity15,18 and demonstrated reduced transactivation of human pituitary gene targets such as the hPRL, hGH and hPit-1 promoters.18 These findings suggested that Arg69 is crucial for PITX2 function. Clinically, all of the affected members bearing the R69H mutation presented with iridogoniodysgenesis.2,17 Furthermore, one affected member showed a broad anterior adhesion of the iris to the posterior periphery of the cornea, obliterating the angle of the anterior chamber in that area.2 These findings approximated to ARS. In our family, the clinical eye features are rather mild (Supplementary Table 1). This probably arises from the different mutational effects of C and H at position 69.
In conclusion, using WES we identified a novel PITX2 mutation, p.R69C, in a family with tooth agenesis associated with clinically inconspicuous IH. WES is a powerful and cost-effective tool to identify the genetic causes of disease.19 In our case, we were able to make the final diagnosis of IH showing obvious tooth agenesis and very mild eye features that we had previously overlooked. WES is particularly useful when clinical diagnosis is difficult or the patients show atypical clinical features.
References
References
Alward WL, Semina EV, Kalenak JW, Heon E, Sheth BP, Stone EM et al. Autosomal dominant iris hypoplasia is caused by a mutation in the Rieger syndrome (RIEG/PITX2) gene. Am J Ophthalmol 1998; 125: 98–100.
Kulak SC, Kozlowski K, Semina EV, Pearce WG, Walter MA . Mutation in the RIEG1 gene in patients with iridogoniodysgenesis syndrome. Hum Mol Genet 1998; 7: 1113–1117.
Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA et al. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet 1996; 14: 392–399.
Shields MB, Buckley E, Klintworth GK, Thresher R . Axenfeld-Rieger syndrome. A spectrum of developmental disorders. Surv Ophthalmol 1985; 29: 387–409.
Heon E, Sheth BP, Kalenak JW, Sunden SL, Streb LM, Taylor CM et al. Linkage of autosomal dominant iris hypoplasia to the region of the Rieger syndrome locus (4q25). Hum Mol Genet 1995; 4: 1435–1439.
Mucchielli ML, Mitsiadis TA, Raffo S, Brunet JF, Proust JP, Goridis C . Mouse Otlx2/RIEG expression in the odontogenic epithelium precedes tooth initiation and requires mesenchyme-derived signals for its maintenance. Dev Biol 1997; 189: 275–284.
Hjalt TA, Semina EV, Amendt BA, Murray JC . The Pitx2 protein in mouse development. Dev Dyn 2000; 218: 195–200.
Lin CR, Kioussi C, O'Connell S, Briata P, Szeto D, Liu F et al. Pitx2 regulates lung asymmetry, cardiac positioning and pituitary and tooth morphogenesis. Nature 1999; 401: 279–282.
Lu MF, Pressman C, Dyer R, Johnson RL, Martin JF . Function of Rieger syndrome gene in left–right asymmetry and craniofacial development. Nature 1999; 401: 276–278.
Kimura M, Machida J, Yamaguchi S, Shibata A, Tatematsu T, Miyachi H et al. Novel nonsense mutation in MSX1 in familial nonsyndromic oligodontia: subcellular localization and role of homeodomain/MH4. Eur J Oral Sci 2014; 122: 15–20.
Kondo Y, Koshimizu E, Megarbane A, Hamanoue H, Okada I, Nishiyama K et al. Whole-exome sequencing identified a homozygous FNBP4 mutation in a family with a condition similar to microphthalmia with limb anomalies. Am J Med Genet A 2013; 161a: 1543–1546.
Arakawa H, Nakamura T, Zhadanov AB, Fidanza V, Yano T, Bullrich F et al. Identification and characterization of the ARP1 gene, a target for the human acute leukemia ALL1 gene. Proc Natl Acad Sci USA 1998; 95: 4573–4578.
Gage PJ, Camper SA . Pituitary homeobox 2, a novel member of the bicoid-related family of homeobox genes, is a potential regulator of anterior structure formation. Hum Mol Genet 1997; 6: 457–464.
Kitamura K, Miura H, Miyagawa-Tomita S, Yanazawa M, Katoh-Fukui Y, Suzuki R et al. Mouse Pitx2 deficiency leads to anomalies of the ventral body wall, heart, extra- and periocular mesoderm and right pulmonary isomerism. Development 1999; 126: 5749–5758.
Kozlowski K, Walter MA . Variation in residual PITX2 activity underlies the phenotypic spectrum of anterior segment developmental disorders. Hum Mol Genet 2000; 9: 2131–2139.
Kissinger CR, Liu BS, Martin-Blanco E, Kornberg TB, Pabo CO . Crystal structure of an engrailed homeodomain-DNA complex at 2.8 A resolution: a framework for understanding homeodomain-DNA interactions. Cell 1990; 63: 579–590.
Chisholm IA, Chudley AE . Autosomal dominant iridogoniodysgenesis with associated somatic anomalies: four-generation family with Rieger’s syndrome. Br J Ophthalmol 1983; 67: 529–534.
Quentien MH, Pitoia F, Gunz G, Guillet MP, Enjalbert A, Pellegrini I . Regulation of prolactin, GH, and Pit-1 gene expression in anterior pituitary by Pitx2: an approach using Pitx2 mutants. Endocrinology 2002; 143: 2839–2851.
Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 2011; 12: 745–755.
Data Citations
Nakashima, Mitsuko HGV Database http://hgv.figshare.com/genome_variation/21 (2014)
Acknowledgements
We thank the patient and her family for participation in this study. We also thank Nobuko Watanabe for her technical assistance. This work was supported by the Ministry of Health, Labour and Welfare of Japan; the Japan Society for the Promotion of Science (a Grant-in-Aid for Scientific Research (B) (25293085, 25293235), a Grant-in-Aid for Young Scientists (B) (25860874), a Grant-in-Aid for Scientific Research (A) (24249019)); the Takeda Science Foundation; the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems; the Strategic Research Program for Brain Sciences (11105137); and a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (12024421).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Human Genome Variation website (http://www.nature.com/hgv)
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Kimura, M., Tokita, Y., Machida, J. et al. A novel PITX2 mutation causing iris hypoplasia. Hum Genome Var 1, 14005 (2014). https://doi.org/10.1038/hgv.2014.5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2014.5
This article is cited by
-
Novel WNT10A variant in a Japanese case of nonsyndromic oligodontia
Human Genome Variation (2023)
-
Pitx genes in development and disease
Cellular and Molecular Life Sciences (2021)
