
Abstract
Cerebellar ataxia (CA) is a disorder associated with impairments in balance, coordination, and gait caused by degeneration of the cerebellum. The mutations associated with CA affect functionally diverse genes; furthermore, the underlying genetic basis of a given CA is unknown in many patients. Exome sequencing has emerged as a cost-effective technology to discover novel genetic mutations, including autosomal recessive CA (ARCA). Five recent studies that describe how exome sequencing performed on a diverse pool of ARCA patients revealed 14 unique mutations in STUB1, a gene that encodes carboxy terminus of Hsp70-interacting protein (CHIP). CHIP mediates protein quality control through chaperone and ubiquitin ligase activities and is implicated in alleviating proteotoxicity in several neurodegenerative diseases. However, these recent studies linking STUB1 mutations to various forms of ataxia are the first indications that CHIP is directly involved in the progression of a human disease. Similar exome-sequencing studies have revealed novel mutations in ubiquitin-related proteins associated with CA and other neurological disorders. This review provides an overview of CA, describes the benefits and limitations of exome sequencing, outlines newly discovered STUB1 mutations, and theorizes on how CHIP and other ubiquitin-related proteins function to prevent neurological deterioration.
Similar content being viewed by others


Introduction
‘Ataxia’ is a general term used to describe a loss of coordination. Ataxia may be caused by a variety of diseases, including metabolic disorders, vitamin deficiencies, peripheral neuropathy, cancer, or brain injuries. However, ataxia may also be the result of progressive deterioration of the cerebellum, which can be caused by a huge variety of relatively rare genetic mutations in a family of disorders termed ‘cerebellar ataxia’ (CA). In addition to alterations in movement and balance, CA diseases can be accompanied by impairments in speech, vision, and cognitive ability. The diseases caused by CA mutations are inherited most commonly in autosomal recessive CA (ARCA, estimated prevalence is 7 per 100,000) or autosomal dominant CA (ADCA, estimated prevalence is 3 per 100,000) manners, in addition to less prevalent mitochondrial or X-linked inheritance. Many forms of ADCAs are caused by polyglutamine expansions within a protein-coding region, whereas some ADCAs and most ARCAs are caused by conventional mutations within the coding region (see Table 1). CAs can also manifest as a secondary feature of neurological diseases affecting the brain, such as Huntington disease, Parkinson disease, cerebral palsy, and dentatorubral pallidoluysian atrophy. The age of onset, prognosis, accompanying symptoms, and possible treatment for a given CA depends on the underlying genetic mutation.
Until a few years ago, the rare occurrence and broad clinical heterogeneity of CAs hindered the identification of underlying genetic contributors. Prior to 2010, it had been estimated that the genetic cause was unknown in ~40% of ADCA and ARCA cases.1 Identifying the genetic basis of CA is crucial for patients because pinpointing the mutation can lead to information on treatments to help manage the disease, identification of relatives currently at risk of developing the disease, and accurate prenatal counseling for family members. Although helping patients is the foremost priority, genetic identification may also implicate novel biological roles for ataxia-associated genes.
The advantages and limitations of using exome sequencing to identify rare disease mutations
As whole-genome sequencing gradually becomes more accessible due to lower costs and better data analysis methods, researchers will continue to learn more about how noncoding regions of the genome may regulate processes in a variety of diseases,2 including neurological diseases.3 However, although the exome (the exon-associated portion of DNA that is transcribed into mature mRNA) represents <1% of the entire genome, the current inventory of disease-causing mutations appear to be disproportionately found in protein-coding regions.4 Exome sequencing has emerged as a powerful technology to identify the genetic cause of rare diseases like CAs, and is emblematic of how specialized technology can be used universally within just a few years. First used successfully to identify the basis of a rare disease in 2009,5 progressively lower costs and faster sequencing platforms have promoted the mainstream use of exome sequencing over whole-genome sequencing today.
Although exome sequencing currently has many advantages over whole-genome sequencing, exome sequencing has technical limitations beyond the inherent inability to detect mutations in noncoding regions, including difficulty in demonstrating sufficient genetic coverage, a potential inadequacy in identifying chromosomal rearrangements, and difficulty in sequencing trinucleotide repeats, such as the polyglutamine repeats, that characterize the majority of known mutations in ADCA. Still, exome sequencing is well suited for studying rare diseases caused by conventional mutations, including ARCAs. There are several approaches a clinical researcher can take when trying to identify the basis of an ARCA. For example, an analysis of the exome profiles of unrelated individuals presenting with the same syndrome may allow researchers to pinpoint a disease-causing mutation. However, a particular CA may be caused by mutations in different genes or by different mutations within the same gene. A second approach to identifying a disease-causing mutation is to sequence exomes from affected and nonaffected individuals within a multigenerational pedigree, although it is not always possible to obtain a sufficient number of genetic samples. In either approach, a major concern in studying autosomal recessive diseases through sequencing is properly filtering single nucleotide polymorphisms against control databases, such as dbSNP and 1000 Genomes Project, since a ‘control’ population has a high likelihood of expressing a recessive mutation.6 In 2011, it was estimated that exome sequencing could identify around 20,000 single nucleotide polymorphisms per genetic sample, of which between 1 and 2% were novel.7 As exome-sequencing data for control populations becomes increasingly accessible,8 it will be easier to differentiate single nucleotide polymorphismss from novel disease-causing mutations.
Identification of a STUB1 mutation associated with Gordon Holmes syndrome
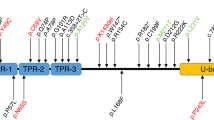
Recently, exome sequencing was used to identify the probable disease-causing mutation in two siblings diagnosed with Gordon Holmes Syndrome (GHS; OMIM #212840), an ARCA characterized by ataxia and hypogonadism.9 The exomes of the two affected sisters and an unaffected male sibling were sequenced, and after filtering for novel variants, a recessive inheritance pattern, location within Identical By Decent regions, and predicted effects on protein function, a single homozygous mutation within the coding region of STUB1 met all criteria in the GHS patients.9 Carboxy terminus of Hsp70-interacting protein (CHIP), the protein product of STUB1, is a cochaperone and ubiquitin ligase that contains three major protein domains: a tetratricopeptide repeat (TPR) domain required for interaction with heat shock proteins (Hsp), a charged domain that mediates CHIP’s dimerization and activity, and a U-box domain that confers ubiquitin ligase activity (Figure 1a). CHIP forms a homodimer and directly binds to Hsp70 and Hsp90 to aid in refolding substrates, or associates with ubiquitin-conjugating enzymes to ubiquitinate substrates with canonical or noncanonical chains.10–14 CHIP can also directly act as an autonomous chaperone by promoting the proper folding and activity of substrates.15,16 The homozygous GHS-associated STUB1 mutation found in this study (c.737C>T) results in the substitution of methionine for threonine at residue 246 (p.T246M) and is located within CHIP’s U-box domain (Figure 1a, Table 2).9 In vitro studies indicated that the T246M mutation abolishes the ubiquitin ligase activity of CHIP but does not disrupt the association between CHIP and Hsp70.9

(a) STUB1 genomic structure and corresponding CHIP protein domains are diagramed. The locations (arrows) of the various mutations associated with ARCA and respective nucleic acid and amino acid changes are indicated in the inset table.9,8–21 Joined arrows indicate a compound heterozygous mutation. (b) The protein structure of RNF216 (left) and OTUD4 (right) are shown with mutations indicated with arrows and identified in the inset table.39,40 The domain abbreviations are: UTR, untranslated region; TPR, tetratricopeptide repeat; CC, coiled coil; RING, really interesting new gene; IBR, in-between RING; OTU, ovarian tumor like.
The GHS patients described in the above study exhibited an unsteady gait that progressed to ataxia, cognitive impairments, and inadequate sexual organ development accompanied by low circulating levels of hormones required for reproductive development (Table 2).9 Interestingly, CHIP−/− mice share these phenotypes, although these deficiencies were initially overlooked due to the more prominent findings of acute stress intolerance and premature aging.11,17 However, on the basis of the data linking CHIP to GHS, the neurological and reproductive phenotypes of CHIP−/− mice were more closely examined, and impairments in motor activity and spatial learning, cerebellar atrophy, and hypogonadism associated with low circulating hormone levels were found.9 The combination of exome sequencing, in vitro data, and recapitulation of the GHS phenotype in a mouse model provide high confidence that CHIP, which was previously unknown to have a direct role in any human disease, is essential for cerebellar maintenance.
Identification of multiple STUB1 mutations associated with ARCA
Bolstering the evidence that loss-of-function mutations in CHIP are causal to ARCA, a second group using exome sequencing reported STUB1 mutations in six patients with ataxia and cerebellar degeneration from three unrelated families.18 In one family, all affected siblings demonstrated a homozygous mutation in STUB1, whereas the patients in the remaining families demonstrated compound heterozygous mutations (Figure 1a; Table 2), all of which were predicted to substantially affect CHIP protein function. The in silico predictions of altered protein function were corroborated with data demonstrating that these various STUB1 mutations were associated with a reduction in the degradation of a known CHIP substrate.18
A third group recently reported novel STUB1 mutations in two siblings with ARCA who demonstrate cognitive deterioration.19 After ruling out polyglutamine expansions and mutations in over a hundred ataxia-related genes, exome sequencing was employed to determine whether a novel mutation was involved. Compound heterozygous mutations in STUB1 were discovered in the two patients; these mutations produced an amino-acid substitution in the charged domain and a deletion in the U-box domain that leads to a frameshift mutation, causing the insertion of eight new amino acids and a premature stop codon (Figure 1a; Table 2).19 The two patients identified in this study developed symptoms in early adulthood that consisted of gait and speech difficulties, cognitive impairment, and cerebellar atrophy, although the circulating levels of testosterone, luteinizing hormone, and follicle stimulating hormone were normal, in contrast to the patients with the T246M mutation.19
In addition, a fourth group performed exome sequencing on a large population of ARCA patients that had been prescreened for mutations in the most common ARCA diseases, and the researchers discovered four novel STUB1 mutations in three patients.20 Two unrelated patients harbored homozygous mutations that led to amino-acid substitutions in the TPR domain and in the U-box domain, whereas the other two patients were siblings and expressed compound heterozygous mutations causing amino-acid substitutions in the same amino acid within the TPR domain (Figure 1a; Table 2).20 Interestingly, all patients in this study had normal levels of estrogen and testosterone.20 The only patient in this study demonstrating cognitive impairment expressed the M240T mutation found within the U-box.20
Most recently, another group has reported on the case of ataxia with myoclonus (muscle jerking), speech difficulties, balance deterioration, and cognitive impairment in a patient harboring compound heterozygous mutations in STUB1.21 One mutation affected the first base of the intron between the fourth and fifth exons, and the other affected the U-box domain.21 Together, these studies demonstrate that mutations in CHIP are causative in multiple ARCA patients from Asian9,18 and Caucasian19–21 ethnicities, and the cerebellar degeneration may or may not be associated with cognitive impairment and hypogonadism.9,18–21 However, judging from the incidence of cognitive impairment occurring in four out of five genetic signatures harboring mutations that affect the U-box (Table 2), it is possible that residual CHIP activity involving an intact TPR domain mitigates clinical symptoms in some patients. At this point, the only available animal model to study CHIP function is the mouse in which the entire STUB1 gene has been deleted.11,17 Although it is difficult to say at this time how mutations within the TRP, charged, or U-box domains specifically affect the symptoms associated with ARCA, the development of animal models with isolated domain mutations may help identify how the bifunctional roles of CHIP affect clinical pathologies.
The pathophysiological role of CHIP in neurodegenerative diseases
Models of neurodegenerative diseases previously identified a plausible role for CHIP in regulating neurological function; however, not until the recent clinical studies has there been any evidence that loss-of-function mutations in CHIP lead to severe CA disease phenotypes in humans.9,18–21 CHIP associates with numerous neuronal proteins. For example, CHIP is detected in Lewy bodies;22 and CHIP recognizes and clears phosphorylated tau23 and α-synuclein.22,24 CHIP suppresses toxicity caused by LRRK225,26 and Huntingtin,27,28 and enhances the ubiquitin ligase activity of wild-type Parkin.29 In a direct link to an ADCA, CHIP mediates the degradation of a mutated polyglutamine-expanded ataxin-1, a causative mutation in spinocerebellar ataxia 1 (SCA1).30 CHIP also participates in a negative feedback cycle with the E2 ubiquitin-conjugating enzyme E2W (Ube2w)and ataxin-3,31 a DUB that targets noncanonical chains.32,33 Similar to ataxin-1, a polyglutamine-expansion mutation in ataxin-3 is implicated in the ADCA SCA3,34 and the severity of the pathological phenotype of transgenic mice expressing the ataxin-3 polyglutamine-repeat mutation is inversely proportional to CHIP copy number expression status.35 It should be noted that the role of CHIP in mitigating mutant ataxin-3 toxicity in other ataxin-3 mutation models is unclear.36 However, it is unlikely that the proteins listed above are implicated in the pathology of the ARCA patients expressing STUB1 mutations, as there were no additional mutations found.9,18–21 Supportive of the notion that loss of CHIP alone directly causes ARCA is the observation that the early-onset of disease and distinctive pathology of ARCA described in these recent studies9,18–21 does not coincide with symptoms of SCA1, SCA3, Parkinson, Alzheimer, or Huntington diseases. Together, these data support a model where CHIP is involved in mediating disease progression when a client protein is mutated; however, it is now clear that a loss of CHIP function, caused by either substitution or truncation mutations (Figure 1a), also leads to a severe pathological consequence through an as yet undetermined mechanism (summarized in Figure 2).

(a) The summary of the neurological proteins that serve as CHIP substrates. (b) The effects of CHIP genetic depletion from mouse models (left) and observed effects on humans with CHIP loss-of-function mutations (right).
Mutations in the ubiquitin ligase RNF216 and the deubiquitianse OTUD4 also associate with GHS
Of the 13 exon mutations in CHIP found in ARCA patients, 9 affect either the charged linker region or the U-box domain (Figure 1a), which are domains required for CHIP function and/or dimerization.37,38 The identification of ataxia-associated mutations that abolish CHIP’s ubiquitination activity,9,18–21 in addition to recent reports of exome mutations in other proteins involved in ubiquitination in other ataxia-related conditions,39,40 support the distinct connection between neurological diseases and defects in the ubiquitin–proteasome system (UPS) (reviewed in references 41–44). A recent study of three siblings with GHS used exome sequencing to find homozygous mutations in both the ubiquitin ligase ring finger protein 216 (RNF216) and the deubiquitinase OTU domain containing 4 (OTUD4), and sequencing of RNF216 in six unrelated GHS patients demonstrated a variety of heterozygous RNF216 mutations (Figure 1b).39 Importantly, the authors verified the involvement of RNF216 and OTUD4 in cerebellar function using zebrafish models demonstrating how the depletion of either gene significantly increases the appearance of cerebellar defects.39 Subsequently, an exome-sequencing study consisting of several ataxia patients identified a unique RNF216 mutation in a GHS patient (Figure 1b).40 Unlike CHIP, targets of RNF216 ubiquitin ligase activity are not well characterized. However, overlap in the GHS phenotype associating with CHIP and RNF216 mutations affords a unique comparative opportunity to study CHIP and RNF216 activities that may provide the molecular starting point to determine the mechanism by which the loss of either E3 ligase could lead to GHS.
CA pathologies—converging at protein quality control?
The diverse etiology of the CA phenotype presents a challenge to researchers. One common feature of some proteins involved in ADCAs is polyglutamine expansion; otherwise, most ADCA proteins are functionally unrelated (Table 1). As discussed above, mutations in different genes such as STUB1 and RNF216 can give rise to a similar phenotype. When the causal genes are functionally related, it may provide some insight into the pathophysiology. However, there are instances where causal genes of a shared phenotype do not appear to be functionally related; for example, Boucher–Neuhäuser Syndrome (OMIM #215470) and GHS are both early-onset ARCAs accompanied by hypogonadotropic hypogonadism. Although Boucher–Neuhäuser Syndrome is characterized by chorioretinal dystrophy and GHS by brisk reflex, both diseases can be caused by mutations in patatin-like phospholipase domain containing 6 (PNPLA6),45 an enzyme with little functional overlap with CHIP or RNF216. It is easy to hypothesize that PNPLA6 is a substrate for either CHIP or RNF216, but there has yet to be any data published to support this, and currently it is unclear how PNPLA6 may intersect with the UPS. However, in addition to the longstanding observation that ubiquitin is present in aggregates that are hallmarks of several neurodegenerative diseases,46 the use of exome sequencing in recent studies demonstrated that multiple components of the UPS are involved in CAs and other neurological and neurodegenerative disorders. For example, ubiquitin carboxyl-terminal esterase L1 (UCHL1), a deubiquitinating enzyme that may also have ubiquitin ligase activity, has previously been linked to Parkinson and Alzheimer diseases.47,48 A homozygous missense mutation in UCHL1 was found in three siblings with ataxia and severe cerebellar atrophy,49 similar to mouse models in which UCHL1 is mutated or deleted.50–52 Similarly, a mutation in ubiquitin protein ligase E3 component N-recognin 4 (UBR4) was detected in a family with episodic ataxia through exome sequencing.53 UPS components have also been found to be mutated in some familial cases of amyotrophic lateral sclerosis, including ubiquilin 2, a ubiquitin-like protein,54,55 and valosin-containing protein (VCP), a ubiquitin segregase.56 Exome sequencing also identified two different mutations in an under-characterized protein, ubiquitin protein ligase E3B (UBE3B), associated with Kaufman Oculocerebrofacial Syndrome,57 whereas other UBE3B mutations result in an intellectual deficiency disorder58 or associate with autism,59 again demonstrating how distinct mutations in the same gene can give rise to different phenotypes. In addition, some ARCAs are attributed to mutations in genes involved in the UPS or in the chaperoning of proteins (Table 1). One only has to look at the myriad known CHIP substrates (Figure 2a) and phenotypes from CHIP loss-of-function studies (Figure 2b) to appreciate the neuronal demand for protein quality control. It is not surprising that a collective theme is emerging, at least within a subset of CAs, that clearly has a direct genetic link to the UPS and protein quality control. Perhaps other known CA casual mutations in genes not directly associated with the UPS cause a change in protein stability, or perhaps participate in signaling networks that intersect the UPS and protein quality control pathways in mechanisms not previously characterized. Future efforts focusing on the molecular characterization of newly identified coding mutations associated with CAs should consider effects on protein stability and possible implications to the UPS, as protein quality control is paramount in maintaining neuronal cell homeostasis.
Conclusions
As the global access to exome-sequencing technology and analysis becomes increasingly available, there is tremendous potential for successful identification of causal mutations of rare diseases, such as CAs. The clinical identification of these mutations, combined with basic and translational research approaches, will foster new insights into human diseases and uncover novel roles for genes and proteins that have direct links to human diseases. We are confident that collaborative studies combining clinical identification of mutations and basic research models validating gene and protein function will uncover the molecular mechanisms by which a single mutation leads to a devastating condition and will foster therapies to help patients with rare genetic diseases.
References
Sailer A, Houlden H . Recent advances in the genetics of cerebellar ataxias. Curr Neurol Neurosci Rep 2012; 12: 227–236.
Ward LD, Kellis M . Interpreting noncoding genetic variation in complex traits and human disease. Nat Biotechnol 2012; 30: 1095–1106.
Ng SY, Lin L, Soh BS, Stanton LW . Long noncoding RNAs in development and disease of the central nervous system. Trends Genet 2013; 29: 461–468.
Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN . The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 2014; 133: 1–9.
Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 2010; 42: 30–35.
Hersheson J, Haworth A, Houlden H . The inherited ataxias: genetic heterogeneity, mutation databases, and future directions in research and clinical diagnostics. Hum Mutat 2012; 33: 1324–1332.
Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 2011; 12: 745–755.
Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012; 491: 56–65.
Shi CH, Schisler JC, Rubel CE, Tan S, Song B, McDonough H et al. Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum Mol Genet 2014; 23: 1013–1024.
Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Höhfeld J et al. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol 2001; 3: 93–96.
Dai Q, Zhang C, Wu Y, McDonough H, Whaley RA, Godfrey V et al. CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J 2003; 22: 5446–5458.
Kampinga HH, Kanon B, Salomons FA, Kabakov AE, Patterson C . Overexpression of the cochaperone CHIP enhances Hsp70-dependent folding activity in mammalian cells. Mol Cell Biol 2003; 23: 4948–4958.
McDonough H, Charles PC, Hilliard EG, Qian SB, Min JN, Portbury A et al. Stress-dependent Daxx-CHIP interaction suppresses the p53 apoptotic program. J Biol Chem 2009; 284: 20649–20659.
Ronnebaum SM, Wu Y, McDonough H, Patterson C . The Ubiquitin Ligase CHIP Prevents SirT6 Degradation through Noncanonical Ubiquitination. Mol Cell Biol 2013; 33: 4461–4472.
Rosser MF, Washburn E, Muchowski PJ, Patterson C, Cyr DM . Chaperone functions of the E3 ubiquitin ligase CHIP. J Biol Chem 2007; 282: 22267–22277.
Schisler JC, Rubel CE, Zhang C, Lockyer P, Cyr DM, Patterson C . CHIP protects against cardiac pressure overload through regulation of AMPK. J Clin Invest 2013; 123: 3588–3599.
Min JN, Whaley RA, Sharpless NE, Lockyer P, Portbury AL, Patterson C . CHIP deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol Cell Biol 2008; 28: 4018–4025.
Shi Y, Wang J, Li JD, Ren H, Guan W, He M et al. Identification of CHIP as a novel causative gene for autosomal recessive cerebellar ataxia. PLoS ONE 2013; 8: e81884.
Depondt C, Donatello S, Simonis N, Rai M, van Heurck R, Abramowicz M et al. Autosomal recessive cerebellar ataxia of adult onset due to STUB1 mutations. Neurology 2014; 82: 1749–1750.
Synofzik M, Schüle R, Schulze M, Gburek-Augustat J, Schweizer R, Schirmacher A et al. Phenotype and frequency of STUB1 mutations: next-generation screenings in Caucasian ataxia and spastic paraplegia cohorts. Orphanet J Rare Dis 2014; 9: 57.
Cordoba M, Rodriguez-Quiroga S, Gatto EM, Alurralde A, Kauffman MA . Ataxia plus myoclonus in a 23-year-old patient due to STUB1 mutations. Neurology 2014; 83: 287–288.
Shin Y, Klucken J, Patterson C, Hyman BT, McLean PJ . The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J Biol Chem 2005; 280: 23727–23734.
Shimura H, Schwartz D, Gygi SP, Kosik KS . CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem 2004; 279: 4869–4876.
Tetzlaff JE, Putcha P, Outeiro TF, Ivanov A, Berezovska O, Hyman BT et al. CHIP targets toxic alpha-Synuclein oligomers for degradation. J Biol Chem 2008; 283: 17962–17968.
Ding X, Goldberg MS . Regulation of LRRK2 stability by the E3 ubiquitin ligase CHIP. PLoS ONE 2009; 4: e5949.
Ko HS, Bailey R, Smith WW, Liu Z, Shin JH, Lee YI et al. CHIP regulates leucine-rich repeat kinase-2 ubiquitination, degradation, and toxicity. Proc Natl Acad Sci USA 2009; 106: 2897–2902.
Jana NR, Dikshit P, Goswami A, Kotliarova S, Murata S, Tanaka K et al. Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J Biol Chem 2005; 280: 11635–11640.
Miller VM, Nelson RF, Gouvion CM, Williams A, Rodriguez-Lebron E, Harper SQ et al. CHIP suppresses polyglutamine aggregation and toxicity in vitro and in vivo. J Neurosci 2005; 25: 9152–9161.
Imai Y, Soda M, Hatakeyama S, Akagi T, Hashikawa T, Nakayama KI et al. CHIP is associated with Parkin, a gene responsible for familial Parkinson's disease, and enhances its ubiquitin ligase activity. Mol Cell 2002; 10: 55–67.
Al-Ramahi I, Lam YC, Chen HK, de Gouyon B, Zhang M, Pérez AM et al. CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J Biol Chem 2006; 281: 26714–26724.
Scaglione KM, Zavodszky E, Todi SV, Patury S, Xu P, Rodríguez-Lebrón E et al. Ube2w and ataxin-3 coordinately regulate the ubiquitin ligase CHIP. Mol Cell 2011; 43: 599–612.
Burnett B, Li F, Pittman RN . The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum Mol Genet 2003; 12: 3195–3205.
Winborn BJ, Travis SM, Todi SV, Scaglione KM, Xu P, Williams AJ et al. The deubiquitinating enzyme ataxin-3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitin chains. J Biol Chem 2008; 283: 26436–26443.
Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet 1994; 8: 221–228.
Williams AJ, Knutson TM, Colomer Gould VF, Paulson HL . In vivo suppression of polyglutamine neurotoxicity by C-terminus of Hsp70-interacting protein (CHIP) supports an aggregation model of pathogenesis. Neurobiol Dis 2009; 33: 342–353.
Tsou WL, Burr AA, Ouyang M, Blount JR, Scaglione KM, Todi SV . Ubiquitination regulates the neuroprotective function of the deubiquitinase ataxin-3 in vivo. J Biol Chem 2013; 288: 34460–34469.
Nikolay R, Wiederkehr T, Rist W, Kramer G, Mayer MP, Bukau B . Dimerization of the human E3 ligase CHIP via a coiled-coil domain is essential for its activity. J Biol Chem 2004; 279: 2673–2678.
Qian SB, Waldron L, Choudhary N, Klevit RE, Chazin WJ, Patterson C . Engineering a ubiquitin ligase reveals conformational flexibility required for ubiquitin transfer. J Biol Chem 2009; 284: 26797–26802.
Margolin DH, Kousi M, Chan YM, Lim ET, Schmahmann JD, Hadjivassiliou M et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med 2013; 368: 1992–2003.
Sawyer SL, Schwartzentruber J, Beaulieu CL, Dyment D, Smith A, Warman Chardon J et al. Exome sequencing as a diagnostic tool for pediatric-onset ataxia. Hum Mutat 2014; 35: 45–49.
Davies JE, Sarkar S, Rubinsztein DC . The ubiquitin proteasome system in Huntington's disease and the spinocerebellar ataxias. BMC Biochem 2007; 8 (Suppl 1 S2.
Duenas AM, Goold R, Giunti P . Molecular pathogenesis of spinocerebellar ataxias. Brain 2006; 129: 1357–1370.
Evert BO, Wullner U, Klockgether T . Cell death in polyglutamine diseases. Cell Tissue Res 2000; 301: 189–204.
Orr HT . Beyond the Qs in the polyglutamine diseases. Genes Dev 2001; 15: 925–932.
Synofzik M, Gonzalez MA, Lourenco CM, Coutelier M, Haack TB, Rebelo A et al. PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain 2014; 137: 69–77.
Alves-Rodrigues A, Gregori L, Figueiredo-Pereira ME . Ubiquitin, cellular inclusions and their role in neurodegeneration. Trends Neurosci 1998; 21: 516–520.
Gong B, Cao Z, Zheng P, Vitolo OV, Liu S, Staniszewski A et al. Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell 2006; 126: 775–788.
Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E et al. The ubiquitin pathway in Parkinson's disease. Nature 1998; 395: 451–452.
Bilguvar K, Tyagi NK, Ozkara C, Tuysuz B, Bakircioglu M, Choi M et al. Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc Natl Acad Sci USA 2013; 110: 3489–3494.
Chen F, Sugiura Y, Myers KG, Liu Y, Lin W . Ubiquitin carboxyl-terminal hydrolase L1 is required for maintaining the structure and function of the neuromuscular junction. Proc Natl Acad Sci USA 2010; 107: 1636–1641.
Saigoh K, Wang YL, Suh JG, Yamanishi T, Sakai Y, Kiyosawa H et al. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat Genet 1999; 23: 47–51.
Yamazaki K, Wakasugi N, Tomita T, Kikuchi T, Mukoyama M, Ando K . Gracile axonal dystrophy (GAD), a new neurological mutant in the mouse. Proc Soc Exp Biol Med 1988; 187: 209–215.
Conroy J, McGettigan P, Murphy R, Webb D, Murphy SM, McCoy B et al. A novel locus for episodic ataxia:UBR4 the likely candidate. Eur J Hum Genet 2013; 22: 505–510.
Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011; 477: 211–215.
Williams KL, Warraich ST, Yang S, Solski JA, Fernando R, Rouleau GA et al. UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol Aging 2012; 33: 2527 e2523–2510.
Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010; 68: 857–864.
Flex E, Ciolfi A, Caputo V, Fodale V, Leoni C, Melis D et al. Loss of function of the E3 ubiquitin-protein ligase UBE3B causes Kaufman oculocerebrofacial syndrome. J Med Genet 2013; 50: 493–499.
Basel-Vanagaite L, Dallapiccola B, Ramirez-Solis R, Segref A, Thiele H, Edwards A et al. Deficiency for the ubiquitin ligase UBE3B in a blepharophimosis-ptosis-intellectual-disability syndrome. Am J Hum Genet 2012; 91: 998–1010.
Chahrour MH, Yu TW, Lim ET, Ataman B, Coulter ME, Hill RS et al. Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet 2012; 8: e1002635.
Acknowledgements
We are grateful to Andrea Portbury for her critical review of this manuscript. This work was supported by the National Institutes of Health grant R01-GM061728 and Foundation Leducq.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/3.0/
About this article

Cite this article
Ronnebaum, S., Patterson, C. & Schisler, J. Emerging evidence of coding mutations in the ubiquitin–proteasome system associated with cerebellar ataxias. Hum Genome Var 1, 14018 (2014). https://doi.org/10.1038/hgv.2014.18
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2014.18
This article is cited by
-
A Chinese Family with Digenic TBP/STUB1 Spinocerebellar Ataxia
The Cerebellum (2024)
-
Spinocerebellar ataxia type 17-digenic TBP/STUB1 disease: neuropathologic features of an autopsied patient
Acta Neuropathologica Communications (2022)
-
Spinocerebellar ataxia type 48: last but not least
Neurological Sciences (2020)
-
Inaugural cognitive decline, late disease onset and novel STUB1 variants in SCAR16
Neurological Sciences (2018)
-
Genetic landscape remodelling in spinocerebellar ataxias: the influence of next-generation sequencing
Journal of Neurology (2015)
