
Abstract
Invasive species have been associated with significant negative impacts in their introduced range often outcompeting native species, yet the long-term evolutionary dynamics of biological invasions are not well understood. Hybridization, either among waves of invasion or between native and introduced populations, could alter the ecological and evolutionary impacts of invasions yet has rarely been studied in marine invasive species. The European green crab (Carcinus maenas) invaded eastern North America twice from northern and southern locations in its native range. Here we examine the frequency of hybridization among these two distinct invasions at locations from New Jersey, USA to Newfoundland, Canada using restriction-site-associated DNA sequencing (RAD-seq), microsatellite loci and cytochrome c oxidase subunit I mitochondrial DNA (mtDNA) sequences. We used Bayesian clustering and hybrid assignment analyses to investigate hybridization between the northern and southern populations. Of the samples analyzed, six locations contained at least one hybrid individual, while two locations were characterized by extensive hybridization, with 95% of individuals collected from Placentia Bay, Newfoundland being hybrids (mostly F2) and 90% of individuals from Kejimkujik, Nova Scotia being classified as hybrids, mostly backcrosses to the northern ecotype. The presence of both F2 hybrids and backcrossed individuals suggests that these hybrids are viable and introgression is occurring between invasions. Our results provide insight into the demographic and evolutionary consequences of hybridization between independent invasions, and will inform the management of green crabs in eastern North America.
Similar content being viewed by others
Introduction
Inter- and intraspecific hybridization in animals has a widely debated role, from being considered an evolutionary ‘dead end’ due to hybrid dysfunction and reproductive barriers, through to having a vital role in creating genetic variability and leading to adaptation and speciation. Hybridization can result in the loss of native genotypes, or lead to an enhanced ability of an invasive species to thrive and expand in its new range (Seehausen, 2013). In addition, hybridization between native and introduced species can lead to the break-up of adaptive gene complexes or replace adaptive alleles over time, and even lead to native extinction through outbreeding depression (Hovick and Whitney, 2014; Todesco et al., 2016). Although hybridization between species can be detrimental and lead to nonviable offspring, this phenomenon may still occur in the absence of reproductive barriers (Abbott et al., 2013; Sedghifar et al., 2016). Hybridization, either between species or genetically divergent populations can lead to adaptive evolution through recombination of parental genotypes, or by the direct introduction of beneficial alleles that can increase fitness (Prentis et al., 2008). In fact, there is growing evidence that hybridization is less a dead end, and likely more often leads to speciation, greater environmental tolerances and release from evolutionary constraints (Burke and Arnold, 2001; Seehausen, 2013; Gramlich et al., 2016). Specifically, the hybridization-invasion hypothesis proposes that hybrids—either intraspecific or interspecific—have enhanced invasiveness or colonization success attributed to the novel phenotypic or genotypic traits hybrids often possess (Hovick and Whitney, 2014; Rius and Darling, 2014). It is therefore apparent that the ability to detect admixed individuals rapidly and accurately has implications in both the conservation of native species and the management of invasive species.
Hybridization studies using genome-wide analyses rather than traditional methods of relatively few genetic markers (for example, microsatellites) can dramatically increase the power to detect hybridization (Twyford and Ennos, 2012). Moreover, the increased numbers of markers as well as genomic coverage is especially beneficial when the (potentially) hybridizing populations or species are closely related, or when the goal is the discernment of more fine-scale hybrid class (Anderson and Thompson, 2002; Vähä and Primmer, 2006; Kovach et al., 2015). Speciation genomics (for example, Feder et al., 2012) is facilitating the understanding of reproductive isolation and local adaptation across the genome using whole-genome sequencing or reduced representation of the genome (Hohenlohe et al., 2010), and surveys of genome-wide single-nucleotide polymorphisms (SNPs) can be useful in studying hybridization between recently diverged lineages providing unprecedented power to identify hybridization (Cristescu, 2015; Viard et al., 2016). This enhanced ability to resolve levels of hybridization in wild populations has clear implications for wildlife management and conservation, yet examples remain rare.
The European green crab (Carcinus maenas) is an invasive species that has rapidly expanded its range across the globe from its native range in Europe and North Africa. In North America, this crab is present on both the Atlantic and Pacific coasts and is continuing to expand both southwards and northwards. On the Atlantic coast, green crabs from a southern European source invaded New York and Massachusetts in the early 1800s, subsequently expanding north to ~45° N. Colonization of higher latitudes only occurred after a second invasion derived from a northern European source occurred in Nova Scotia in the 1980s (Audet et al., 2003; Roman, 2006). The current genetic structure of green crabs in the northwest Atlantic appears to mirror that of their native Europe, and physiological evidence supports that these crabs are locally adapted to these northern and southern thermal regions along their invasive range (Tepolt and Somero, 2014; Tepolt, 2014). Previous genetic studies support two genetically distinct populations now ranging from Virginia, USA, to Newfoundland, Canada with an approximate contact zone in southern Nova Scotia (Blakeslee et al., 2010; Pringle et al., 2011; Darling et al., 2014; Tepolt and Palumbi, 2015). Recently, Jeffery et al. (in press) revealed a surprising amount of genomic differentiation between the populations along the eastern coast of North America derived from the two invasions (average FST between northern and southern populations based on SNPs=0.11), warranting the designation of two distinct ecotypes that are spatially separated, but with a contact zone in southern Nova Scotia. Moreover, preliminary evidence suggests an independent hybridization event between these divergent populations in Placentia Bay, Newfoundland, likely derived from an admixed source population from a known admixture zone in Nova Scotia in the early 2000s (Blakeslee et al., 2010; Darling et al., 2014; Tepolt and Palumbi, 2015). The high degree of propagule pressure is suggested to explain the green crab’s range expansion and ongoing intraspecific hybridization in this region (Rius and Darling, 2014). Admixture between the northern and southern populations of green crab in their invasive range(s) may in time lead to evolution and adaptive radiations to differences in environmental tolerances, making the future spread and ecological impact of such hybrid crabs difficult to predict (Seehausen, 2013; Rius and Darling, 2014). Viable hybrid populations of green crab suggest that current assessments that treat green crab as a single ecotype (Therriault et al., 2008) may be inadequate to fully understand or predict demographic trajectories of green crab in the northeastern Atlantic.
The overall objective of this study was to quantify the presence and geographic extent of hybridization across populations of invasive green crab along a latitudinal gradient in eastern North America and to explore what role, if any, hybridization may have in the future expansion of their invasive range. We aimed to first compare evidence for hybridization using a large panel of SNPs obtained by RAD sequencing, as well as 13 microsatellite loci and mitochondrial cytochrome c oxidase subunit I (COI) sequences for 11 sampling sites and to compare the ability of these different markers to detect hybridization. Second, we aimed to (i) determine the specific composition of hybrid classes at each location, and (ii) to estimate the direction of gene flow among populations. Our results explore the evolutionary consequences of multiple invasions, and specifically for green crab, highlight the implications of hybridization between two independent invasions potentially adapted to different temperature regimes.
Materials and methods
DNA extraction
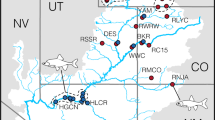
Green crabs were collected from 11 sites ranging from New Jersey, USA, to Newfoundland, Canada (Table 1 and Figure 1). All the sites were sampled in 2011, except New Hampshire, which was sampled in 2013. The tissue samples were collected and preserved in AllProtect (Qiagen, Toronto, ON, Canada) or 80% ethanol. DNA was isolated from tissue samples using phenol:chloroform extraction or NucleoMag 96 Tissue (Macherey-Nagel, Bethlehem, PA, USA) following the manufacturer’s protocol, including RNase A (Qiagen) treatment. All DNA samples were quantified using the Qubit dsDNA HS Assay Kit (Life Technologies, Burlington, ON, Canada) with the assays being read on a Qubit v2.0 (Life Technologies) or Quant-iT PicoGreen dsDNA Assay Kit (Life Technologies) with the assays being read on a FLUOStar OPTIMA fluorescence plate reader (BMG Labtech, Ortenberg, Germany).

Bathymetry map showing the 11 sampling locations for green crab (Carcinus maenas) in eastern North America, ranging from New Jersey to southern Newfoundland. Approximate locations of the initial northern and southern invasions are marked with asterisks (*). The 1000 m isobath line is highlighted offshore for reference.
mtDNA analysis
A 502 base pair fragment of the mitochondrial cytochrome c oxidase subunit I (COI) gene was amplified using M13F (-21)/M13R (-27) tagged version of the primers described in Roman and Palumbi (2004) (M13F-CmaCOIF 5′-TGTAAAACGACGGCCAGTGCTTGAGCTGGCATAGTAGG-3′ and M13R-CmaCOIR 5′-CAGGAAACAGCTATGACGAATGAGGTGTTTAGATTTCG-3′; Integrated DNA Technologies, Coralville, IA, USA). PCR reactions were carried out in 50 μl volumes comprising 100 ng DNA, 1X AmpliTaq 360 Buffer (Life Technologies), 1.5 mm MgCl2, 0.2 mm each nucleotide (dNTP), 0.5 μm each primer and 0.025 U AmpliTaq 360 (Life Technologies). Thermal cycling conditions consisted of 94 °C for 3 min; 30 cycles of 94 °C for 30 s, 50 °C for 30 s and 72 °C for 30 s; 1 cycle of 72 °C for 10 min. To verify amplification, 5 μl of the PCR product was electrophoresed on a 1% agarose gel, visualized using SYBR Safe (Life Technologies) and documented using a Gel Logic 200 (Kodak, Rochester, NY, USA). Forty microliters of the remaining PCR product was purified using QIAquick PCR purification kit (Qiagen). The purified PCR products were quantified as described above and sequenced in both directions using M13F (-21) and M13R (-27) and BigDye Terminator v3.1 cycle sequencing kit (Life Technologies) per the manufacturer’s instructions with the following modification, 0.5 μl of the BigDye Terminator v3.1 Ready Reaction Mix (Life Technologies) and 3.8 μl of the 5X sequencing buffer (Life Technologies) was used per 20 μl reaction. Sequencing products were size separated on a 36 cm array containing POP7 polymer on an AB3130xl genetic analyzer (Life Technologies).
The sequencing results from both directions were assembled using Vector NTI Advance v11 (Life Technologies). All sequences were free of indels and trimmed to the same length of 481 bp. For identification, the resulting sequence for each individual was then aligned to reference sequences, haplotypes H1–H6 and H8–H10 from Darling et al. (2008) (Genbank accession numbers FJ159008-FJ15016) using Vector NTI Advance v11 (Life Technologies).
RAD sequencing
Restriction-site-associated DNA sequencing (RAD sequencing) libraries were prepared as described by Etter et al. (2011b) (see also Etter et al., 2011a) with modifications described in detail in Jeffery et al. (in press). Only DNA samples with a visible, high molecular weight band on the agarose gel and positive for an M13F-CmaCOIF/ M13R-CmaCOIR amplification product were used for library preparation. Each library comprised DNA samples from 22 individuals (1 μg DNA per individual) from the same geographical location with each individual sample having a different in-line barcode in the P1 adaptor. Amplification cycles for all libraries were 98 °C for 30 s; 14 cycles of 98 °C for 30 s, 65 °C for 30 s, 72 °C for 30 s; 1 cycle of 72 °C for 5 min. All libraries were sequenced on a HiSeq 2000 (Illumina, San Diego, CA, USA) as 100 bp paired-end sequences with one library per lane. Sequencing was performed at the McGill University and Génome Québec Innovation Centre, Montréal, Canada.
RAD data for the 242 individuals collected for this study were processed in Stacks v.1.21 (Catchen et al., 2013) where one individual was removed due to low sequence coverage. The ustacks module was used to create stacks of 80 bp RAD tags with a minimum depth of 5 and a maximum distance of 4 between stacks. We used cstacks to construct a catalog of 46 067 loci based on sequence identity. These were then filtered in the Stacks populations module to include RAD tags only present in >75% of individuals and a minor allele frequency greater than 5%, retaining 15 847 loci. This data set was then filtered for departures from Hardy–Weinberg equilibrium and missing data, where individuals missing 25% of loci or loci missing >5% of genotypes were removed using PLINK (Purcell et al., 2007).
Microsatellite analysis
A total of 575 individuals were genotyped at 13 microsatellite loci using three multiplex panels (see Supplementary Table S1). PCR reactions were performed in 10 μl volumes containing 10 ng DNA, 1X Type-it Multiplex PCR Master Mix (Qiagen, Toronto, Canada), and 0.1–1.0 μm each primer (see Supplementary Table S1 for panel construction and primer volumes). The PCR cycles consisted of an initial denaturation step of 95 °C for 5 min followed by 28 cycles of 95 °C for 30 s, annealing temperature for 5 min and 72 °C for 30 s; this was followed by one cycle of 60 °C for 30 min. The annealing temperatures for the three panels were: CMAP01 and CMAP03 annealing temperature =58 °C X=90 s; CMAP02 annealing temperature =55 °C X=3 min. The PCR products were size fractionated on an AB3130xl capillary electrophoresis system (Life Technologies) using a 36 cm array and POP7 polymer. An internal size standard, GeneScan 500 LIZ (Life Technologies), was included with each sample for the size fractionation. The resulting electropherograms were analyzed using GeneMapper version 5.0 (Life Technologies) to determine the relative (to the size standard) allele sizes. During sample analysis, two types of controls (intra- and inter-plate controls) were included on each 96-well plate of DNA samples. These samples were used to identify errors made during sample processing (such as a plate being reversed, or a row of samples duplicated or missed).
Population structure and hybrid detection
To explore the power of the Bayesian clustering programs NEWHYBRIDS (Anderson and Thompson, 2002) and STRUCTURE (Pritchard et al., 2000) to identify hybrids and hybrid classes, we first identified pure north and pure south individuals on the basis of their membership coefficients (Q-values) in preliminary STRUCTURE analyses using the full RAD and microsatellite panels. We used the R package ParallelStructure (Besnier and Glover, 2013) to run STRUCTURE v2.3.4 (Pritchard et al., 2000) to identify population structuring among all sampled localities, and to estimate the best value of K, the number of putative populations. We used the admixture model with 100 000 burn-in iterations and 500 000 Markov Chain Monte Carlo steps with default parameters and no prior structuring information. We let K range from 1 to 11, and replicated each run three times. We then evaluated the best estimate of K using STRUCTURE HARVESTER (Earl and vonHoldt, 2012) and CLUMPAK (Kopelman et al., 2015) using the delta K statistic described by Evanno et al. (2005).
Next, we created simulated data sets of each of six genotype classes using 50 pure northern and pure southern individuals whose Q-values were >0.90 or <0.10, respectively using the R packages hybriddetective (Wringe et al., in review; available at https://github.com/bwringe/hybriddetective) and parallelnewhybrid (Wringe et al., 2017). Simulated hybrids were constructed separately using the 13 microsatellite loci and a subset of 200 diagnostic SNP loci selected using the getTopLoc function from hybriddetective, which returns a desired number of loci by their FST using the package hierfstat (Goudet, 2005) and then selects the top user-specified number of unlinked loci using PLINK (Purcell et al., 2007). These diagnostic loci were subset from the full RAD panel and converted to NEWHYBRIDS format using the R package genepopedit (Stanley et al., 2017). The package parallelnewhybrid then runs NEWHYBRIDS in parallel, allowing multiple data sets and replicates to be analyzed quickly. The simulated data created for both microsatellite and filtered SNP loci panels were then run through STRUCTURE using the above parameters and NEWHYBRIDS to determine the efficacy of these programs in detecting hybrid individuals.
We implemented NEWHYBRIDS v.1.1 (Anderson and Thompson, 2002), which applies a Gibbs sampler and Markov Chain Monte Carlo to estimate posterior probabilities of each individual’s membership as a pure parent, first- or second-generation hybrid (F1 or F2), or an F1 backcross with a pure parent. NEWHYBRIDS was run in parallel using parallelnewhybrid (Wringe et al., 2017). We then ran our microsatellite data and 200 high FST/low linkage diagnostic SNP panels through NEWHYBRIDS. The simulated pure populations were included in these runs with the real population data using the ‘z’ and ‘s’ designations indicating the pure northern or southern genotypes. We used Jeffreys prior probabilities and default genotype proportions, and the Markov chain was left to run for 300 000 sweeps following a burn-in of 50 000 iterations. We also calculated hybrid indices for individuals in suspected hybrid populations in GENODIVE (Meirmans and Van Tienderen, 2004) using the maximum-likelihood method of Buerkle (2005). We grouped the northern populations and southern sites into two pure north and south groups after removing individuals with intermediate Q-values, with a third group comprising KJI (Kejimkujik, NS, Canada) and PLB (Placentia Bay, NL, Canada) tested for admixture. In this instance, hybrid indices closer to 1.0 means individuals have a close affinity with the northern population, while an index closer to 0 means a closer affinity with the southern population.
Finally, a COI minimum spanning haplotype network was constructed using PopArt 1.7 (Leigh and Bryant, 2015) with each haplotype showing relative proportions of northern or southern individuals.
Demographic inference
We explored different two-population demographic scenarios using ∂a∂i (diffusion approximation for demographic inference; Gutenkunst et al., 2009) to estimate the approximate divergence time between the ancestral northern and southern populations of crabs. This software uses an approximation to the allele frequency spectrum of the data to produce a composite log-likelihood value for each model tested. We used a panel developed by Jeffery et al., (in press) of 8326 putatively neutral SNPs characterized by low linkage disequilibrium, to test five population dynamic models, including a strict isolation model, an ancient migration model, a secondary contact model, a model with an ancient population bottleneck followed by growth and a split, and a split with migration model (Supplementary Figure S1). Parameters were optimized for each model three times with different initial parameters using two different optimization functions included in the ∂a∂i software. We then estimated the time the two populations split (T) based on an optimal value of Θ calculated in ∂a∂i, where Θ=4Nrefμ, with μ=10−9 based on the crustacean Daphnia’s nuclear mutation rate (Xu et al., 2012), Nref is the reference ancestral effective population size and T=2Nref × 3 years, the minimum generation time of C. maenas (Berrill, 1982). In addition, we tested four three-population models with different migration rates and levels of admixture to account for the demographic process among northern, southern and admixed populations.
To test for the presence and directionality of gene flow among sampling sites, we used TREEMIX 1.12 (Pickrell and Pritchard, 2012) to build maximum-likelihood trees of the full RAD SNP panel with all sampling sites. Files were converted to allele frequencies per population using genepopedit (Stanley et al., 2017) and a Python script included in TREEMIX. We then tested a range of 1–10 migration events and selected the most likely number of migration events based on the log-likelihood of the event and the plotted residuals. The trees and migration events were plotted in R using customized functions based on the functions included in TREEMIX. We also used a three-population approach (Reich et al., 2009) using the THREEPOP module contained within TREEMIX. This approach calculates an f3 statistic for all possible triplets of populations in the form of (A:B,C), where a significantly negative f3 value indicates that A is a hybrid of B and C. Although the three-population test provides evidence for admixture with a negative f3 statistic, positive f3 values do not necessarily reject admixture (Reich et al., 2009). We used random subsets of 500 SNPs as recommended by the program authors to calculate the standard error of the f3 statistics for the SNP panel. Finally, we examined the presence and directionality of migration among sites using the divMigrate() function in the R package diveRsity (Keenan et al., 2013).
Results
Genetic markers
Our final data set consisted of trimmed 481 bp sequences of the mitochondrial COI gene for 260 individuals, 13 microsatellite loci for 575 individuals and 9137 SNP loci for 241 individuals from across 11 sampling sites on the coast of eastern North America (Table 1 and Figure 1). For simulations and analyses in STRUCTURE and NEWHYBRIDS, we used a panel of 200 high FST unlinked loci created with hybriddetective in R from our full RAD panel. These 200 SNPs were chosen from the full panel of available SNPs to minimize both linkage disequilibrium among SNPs and the computational time required for each NEWHYBRIDS run (Anderson and Thompson, 2002).
Hybrid detection power
The efficacy of the 200 diagnostic SNPs from our RAD panel and 13 microsatellite loci for hybrid detection were each tested using three independently simulated multigenerational hybrid data sets replicated three times each and analyzed in NEWHYBRIDS. The top 200 SNPs allowed correct assignment of an individual as a pure northern, pure southern or hybrid individual 98–100% of the time at all posterior probability thresholds (Supplementary Figure S2). Of the six possible genotype classes, mean hybrid assignment success ranged from 96% for F2 hybrids to 98–100% for pure populations at a probability threshold of 0.99 (Supplementary Figure S3). All genotype classes except F2s were assigned with 100% success at probability thresholds of 0.9. The performance of the 13 microsatellites was markedly worse than that of the SNPs, especially in terms of correctly assigning pure northern or southern individuals (Supplementary Figure S4). At probability thresholds of 0.99, there was 0% assignment success to a pure northern or southern population, whereas hybrid assignment was 82% at a threshold of 0.99. However, when divided into specific hybrid genotype classes, assignment success declined and ranged from ~40 to 85% at probability thresholds of 0.5, but dropped to 0% at thresholds of 0.99 for all pure and hybrid classes (Supplementary Figure S5). Overall detection of hybrids regardless of specific hybrid class was highly correlated between simulated microsatellite and RAD data sets (r=0.98, P<0.005); however, due to the reduced ability of microsatellites to detect specific hybrid classes at high probabilities, we based our overall hybrid detection results on the top 200 SNPs only.
We then determined the ability of STRUCTURE to detect admixed individuals from our microsatellite and diagnostic SNP panels using the simulated data sets. STRUCTURE analyses on the simulated diagnostic SNP data created with hybriddetective in R showed expected admixture proportions for each genotype class, as was the objective, and the delta K statistic calculated in CLUMPAK and Structure Harvester provided evidence for K=2. Simulated microsatellite data run through STRUCTURE also provided evidence for K=2 (Supplementary Figure S6), though expected admixture proportions for F1 and F2 hybrids were less apparent than when using diagnostic SNPs.
Population structuring and hybrid assignment
Mitochondrial COI haplotype diversity was highest in KJI and PLB, with five haplotypes each, and between two to four at each other site. The five most abundant COI haplotypes were represented by individuals from both northern and southern populations, suggesting potential introgression of mitochondrial haplotypes into hybrid individuals (Figure 2a, Supplementary Figure S7). STRUCTURE analyses on the individuals using both RAD-seq SNPs and microsatellites indicated that our sampling locations were separated into northern and southern populations based on the delta K statistic of Evanno et al. (2005) (Figures 2b and c). Delineation between these populations corresponds to the northern limits of the initial North American invasion and the southern limit of the more recent secondary invasion in Nova Scotia from a northern European source. STRUCTURE results indicated hybrid individuals with intermediate Q-values at KJI and PLB, though these could not individually be categorized to hybrid class within STRUCTURE itself (Figure 2 and Supplementary Figure S6). Admixture coefficients showed a skew towards the northern population, consistent with northward gene flow (Supplementary Figure S8). NEWHYBRIDS analysis using the SNP panel indicated that both KJI and PLB consisted almost entirely of crabs of hybrid origin between the northern and southern populations (90% and 95%, respectively), though with different overall compositions (Figures 3 and 4). KJI mostly comprised backcrossed individuals to the pure northern population (76%) with some F2 hybrids (10%). PLB mainly comprised F2 hybrids (60%) with 9% backcrossed individuals to the pure southern population, and 14% of individuals showing high probability of being backcrossed to the pure northern population. Evidence of hybridization was also observed in five other locations (BDB, CBI, CLH, MBO and TKT) where hybrids were found to make up between 5 to 32% of the individuals studied (Figure 3). Though the microsatellites could not resolve individuals to specific hybrid classes, both SNPs and microsatellites consistently identified the same individuals as hybrid (regardless of class) in KJI and PLB (r=0.85, P<0.001).

Genetic and genomic differences among populations of green crab. (a) Mitochondrial COI haplotypes detected at each population. Populations with extensive hybridization (KJI and PLB) contain haplotypes common to both the northern and southern populations. Haplotypes are named following Darling et al. (2008). (b and c) STRUCTURE results on actual data from the microsatellites (b), and the full RAD panel (c). The best value of K, the putative number of populations based on the delta K method of Evanno et al. (2005), is equal to two in both cases.

Hybrid class analysis of populations using microsatellites and SNPs. (a) Microsatellite results from NEWHYBRIDS, where individuals were classed as either a pure northern or southern population individual, or a hybrid. (b) Results from NEWHYBRIDS for 200 diagnostic SNPs filtered for high FST and low linkage. Individuals were classed as one of six possible genotypes: pure north or pure south population, F1 or F2 hybrids, or backcrosses to either of the pure populations. Evidence of hybridization is seen primarily at KJI and PLB for both data sets. SN and SS denote simulated northern and simulated southern individuals, which were included in each NEWHYBRIDS run.

The proportion of pure northern, pure southern, and hybrid individuals based on NEWHYBRIDS genotype classification at each location.
Maximum-likelihood hybrid indices from GENODIVE for KJI and PLB using the full RAD panel ranged from 0.60 to 0.91 and from 0.27 to 0.82, respectively (Figure 5). These are consistent with results from NEWHYBRIDS, as the higher hybrid index values for KJI suggest a closer affinity to the northern population consistent with what would be expected of backcrosses, while the more intermediate values at PLB congruent with a higher proportion of F1 or F2 hybrids rather than pure or backcrossed individuals.

Hybrid indices for admixed populations. (a) Frequency distribution of maximum-likelihood hybrid indices for individuals from admixed sites, including Kejimkujik (KJI) and Placentia Bay (PLB) calculated in GENODIVE. (b) Individual hybrid indices with 95% confidence intervals for each individual from KJI and PLB.
Demographic inference
Of the five two-population models tested in ∂a∂i, the strict isolation model best suited our data when it was split into two populations with admixed individuals removed, yielding the best log-likelihood (−5400.12) and smallest residuals (Supplementary Table S2 and Supplementary Figure S9). Using a nuclear mutation rate of 10−9 (Xu et al., 2012) and a population mutation rate (Θ) of 2820.19 calculated in ∂a∂i, we estimate that the ancestral northern and southern source populations of green crab diverged approximately 470 000 years ago.
A maximum-likelihood (ln likelihood =525.197) tree with estimated migration rates yielded separate northern and southern population clusters, with the exception that PLB clustered with the southern populations (Figure 6). The allele frequency covariance matrix residuals were lowest overall for nine migration events (Supplementary Figure S10). TREEMIX indicated migration to PLB from both northern and southern sites, and a migration event from the southern population towards KJI, consistent with secondary contact at this region. There were five additional migration events included in the tree from south to north but these were not weighted as heavily as the events leading to the admixed populations (Figure 6). The top 10 most significant three-population tests also provided evidence of admixture of a northern and southern site occurring in the three northern sites KJI, PLB and MBO (Supplementary Table S3). The f3 statistics were similar among each three-population combination, and each was significant at a z-score <−20. We verified the presence of gene flow to PLB from both northern and southern locations using diveRsity, which indicated numerous migration events to PLB from both the northern and southern populations, as well as migration within sites categorized as north or south, yet no migration events from the south to KJI (Figure 7). Admixture and migration from both northern and southern populations into KJI is unsurprising given it is geographically intermediate to the northern and southern populations.

Maximum-likelihood phylogram constructed in TREEMIX for the full RAD panel of SNPs with nine supported migration events. Events with the greatest relative weight tend to move towards hybrid populations KJI and PLB. The drift parameter represents the degree to which genetic drift has occurred between each population.

Relative strength and direction of migration among locations using divMigrate (Sundqvist et al., 2016). The legend shows relative migration rates, where darker arrows indicate higher levels of gene flow.
Discussion
Hybridization is an interesting biological phenomenon, which has impacts ranging from hybrid inviability to the evolution of adaptation and increased fitness (Burke and Arnold, 2001). Here we investigated intraspecific hybridization between two invasions of green crab that independently invaded the east coast of North America in the 1800s and again in the 1980s. We build on previous works identifying a secondary contact zone among invasion waves in southern Nova Scotia and the presence of hybridization (Blakeslee et al., 2010; Pringle et al., 2011; Darling et al., 2014; Tepolt and Palumbi, 2015). We revealed that six of 11 locations sampled contained evidence of hybridization and that two locations in particular, Kejimkujik, Nova Scotia (KJI) and Placentia Bay, Newfoundland (PLB) showed particularly high levels of introgression. These populations consisted of 90% and 95% hybrid individuals, respectively, of which PLB primarily consists of F2 hybrids, and KJI consists of backcrossed hybrids. The long-term consequences of introgression remain unknown, but continued monitoring of green crab populations will be necessary to determine any impacts on this hybridization process on their ability to expand and colonize new environments.
Hybridization in Atlantic marine animals
Hybridization, whether intra- or interspecific, has frequently been shown to lead to increased colonization invasion success in both plants and animals (Hovick and Whitney, 2014; Rius and Darling, 2014). Interspecific hybridization in the genus Carcinus (C. maenas X C. aestuarii) has previously been implicated in facilitating the adaptive change that allowed these hybrid crabs to invade and become established along the Japanese coast (Darling, 2011). Although preliminary evidence of intraspecific hybridization has been noted within green crabs (C. maenas) along the North American invasive range (Blakeslee et al., 2010; Darling et al., 2014), our study is the first to directly quantify the composition and geographic extent of hybrids relative to the pure parental populations on a genome-wide scale, integrating SNPs, microsatellites and mitochondrial sequences.
Numerous recent studies have highlighted the ability of highly diagnostic SNPs in revealing intraspecific hybridization between ecotypes that have undergone long-term isolation with recent secondary contact. For example, Le Moan et al. (2016) revealed hybridization between previously isolated coastal and offshore ecotypes of European anchovy (Engraulis encrasicolus) in the Atlantic and Mediterranean using a small panel of 300 diagnostic SNPs derived from a RAD-seq catalog of 5638 SNPs. Bradbury et al. (2014) revealed significant hybridization but little introgression (that is, few F2 or later-generation hybrids detected) among co-occurring populations of genetically distinct Atlantic cod (Gadus morhua). Finding few later-generation hybrids led Bradbury et al. (2014) to suggest strong selection against hybrids or genetic incompatibility of hybrid individuals prevents gene flow among these populations. In contrast to the Atlantic cod and anchovy, adult green crab movement is limited and the majority of dispersal is likely achieved during the planktonic larval stages. The range of green crab and any genetic or population components will therefore mainly depend on prevailing currents in addition to gradual movement up or down the coastline or human introduction as is likely the case in Newfoundland. The northern and southern population structuring in North America appears to reflect historical structure of the source populations in northern and southern Europe (Roman, 2006; Darling et al., 2008; Tepolt and Palumbi, 2015). Hybridization is then highly influenced by natural dispersal and anthropogenic sources promoting secondary contact between these ecotypes at various points along their range. Although our study suggests that first-generation hybrid green crabs are indeed viable, previous work suggests significant divergence of these populations visible in genome-wide differences observed between the northern and southern populations (Jeffery et al., in press). The consequences of the observed hybridization could be substantial if hybrid crabs are suited to invade new habitats relative to their parental ecotypes, necessitating further monitoring and investigation of hybrid populations.
Marker comparison for hybrid quantification
All three marker types (mitochondrial DNA, microsatellites and SNPs) showed clear support for hybridization at KJI and PLB. COI haplotype diversity was highest at these sites, with five haplotypes each, whereas only two to four haplotypes were found at each of the sites considered as pure northern or southern populations, corroborating previous work (Blakeslee et al., 2010). Although this does not necessarily mean that hybridization has occurred, as COI is typically a maternally inherited haploid marker, it does provide evidence for the presence of individual crabs from both northern and southern populations within the admixed populations, allowing the possibility of hybridization. Bayesian clustering analyses on both the microsatellite and SNP data showed clear evidence of hybridization at PLB and KJI, though the SNP panel was better able to resolve the memberships of individuals to the northern or southern populations than the microsatellites. Hybrid detection power varied between the simulated microsatellite and filtered SNP data, and was considerably higher using the unlinked high FST diagnostic SNPs. Hybrid power analyses suggest that our 13 microsatellites can never achieve 100% assignment success to hybrid class at a posterior probability of 0.99. In fact, assignment success was always below 90% at a probability of 0.5 and dropped to 0% at a probability of 0.99 for all genotype classes. In STRUCTURE, microsatellites were able to reliably distinguish pure parental populations, but were worse at distinguishing hybrids; this is opposite to the pattern found using NewHybrids, where microsatellites were better able to distinguish hybrids overall. However, when separating ‘hybrids’ into their respective classes, NewHybrids was better able to distinguish parental populations from F1, F2 and backcross hybrids. This distinction between marker types may be due to the different numbers of markers used, as we only genotyped 13 microsatellites but were able to filter thousands of SNPs to select the most diagnostic markers. Randi (2008) recommends the use of simulated genotype data to ascertain hybrid membership reliability for the markers used in a given study, and the use of >20 unlinked microsatellite loci or larger numbers of SNP loci to accurately detect F1, F2 and first-generation hybrid backcrossed individuals. This reinforces the utility of large genome-wide SNP panels to infer hybrid class. Here we demonstrate that a panel of a few hundred diagnostic SNPs can be used to resolve hybrid class.
Population structure and hybrid assignment
Perhaps most importantly, we have revealed the wide geographic extent of hybridization in the northwest Atlantic green crab invasive range, and extensive hybridization at two sites of known secondary contact. These sites could represent hybrid swarms (sensu Harrison, 1993), where secondary contact between parental populations, and a lack of reproductive isolation, has facilitated a successful second generation of hybrids (F2s and backcrosses) to occur. The distribution of the hybrids is consistent with KJI as a natural secondary contact zone between the northern and southern populations, and these ecotypes likely correspond to the northern and southern ecotypes in the native European range (Roman, 2006). Native populations show a genetic continuum based on COI and microsatellite data, which suggests high levels of gene flow, but adaptive differences at each extreme of this latitudinal cline based on genome-wide SNPs (Roman and Palumbi, 2004; Darling et al., 2008; Tepolt and Palumbi, 2015). Interestingly, we found greater evidence of crabs in KJI backcrossing to individuals from the northern population than to the southern population. This is perhaps consistent with northern larvae being transported to KJI by the prevailing southwestern current along the Scotian shelf allowing for the backcross to occur (Pringle et al., 2011). In contrast, PLB is geographically isolated from the southern population, and so the hybridization observed is likely the result of introduction from a previously admixed Scotian shelf population (Blakeslee et al., 2010) or possibly multiple anthropogenic introductions from separate northern and southern populations. Our results conform to the former hypothesis as we observed only one individual in this location that is considered of ‘pure’ northern origin, suggesting the introduction of already-admixed individuals to this location rather than separate introductions of northern and southern individuals.
Placentia Bay experiences a heavy volume of commercial and recreational ship traffic, which are the most likely causes for the introduction of green crabs to Newfoundland, as opposed to the prevailing currents, which travel in the opposite direction (Blakeslee et al., 2010; Pringle et al., 2011). Correspondingly, three-population tests (Reich et al., 2009) showed significant admixture at a number of sampling sites, the most admixed of which were KJI and PLB. For these sites, evidence of admixture was always between a northern and southern site; this, however, does not necessarily mean that the populations at these sites are actually results of admixture between each northern site with a southern site, but rather provides evidence of a lack of ‘treeness’ that overall suggests admixture between at least one northern and one southern site (Pickrell and Pritchard, 2012). Consistent with previous studies (for example, Darling et al., 2014), the presence of second-generation (F2) hybrids and backcrosses suggests that hybrid offspring of the northern and southern ecotypes of green crab in eastern North America are viable.
We suggest that hybridization is occurring in these regions simply due to secondary contact between ecotypes and a lack of reproductive barriers, which would prevent fertilization (pre-zygotic) or the production of viable offspring (post-zygotic). An alternative hypothesis to natural secondary contact is the hybridization-invasion hypothesis, whereby hybridization among lineages leads to increased genotypic and/or phenotypic variation that allows for the ability to colonize new environments and then persist in that environment (Hovick and Whitney, 2014). The fact that such high proportions of hybrid individuals are found at KJI and PLB may lend support to an inherent hybrid advantage at these locations. For example, hybrid advantage has been equated to broader environmental tolerances and-or increased fecundities relative to either pure form (Rius and Darling, 2014); however, this is speculative and hybrids may in fact have only intermediate thermal tolerances relative to either parental lineage, which tend to be adapted to warm or cold waters (Tepolt and Somero, 2014). The phenomenon of hybrid advantage is well documented and is known to occur among natural populations (Gramlich et al., 2016) and to select for desired traits in the aquaculture trade (for example, Zheng et al., 2011). In fact, hybridization is often observed to facilitate the invasion and establishment of non-native species due to hybrid advantage and potential speciation (Lee, 2002; Facon et al., 2005). To test this hypothesis, further investigations and experimental evidence of the relationships between green crab ecotype distribution and changes in environmental parameters along their invasive range will be required.
Demographic inference and migration
Inferences of both the historical divergence between these lineages and levels of contemporary gene flow can be made using our SNP data. Based on the population mutation rate in a strict isolation demographic model, we find that the source populations of green crabs split approximately 470K years ago, suggesting long-term isolation of these ecotypes due to adaptive differences in environmental tolerance that has then carried over into its invasive range. In the native range, green crab show a distinct latitudinal cline in mitochondrial COI haplotypes but relatively little latitudinal differentiation in microsatellites, which suggests a genetic continuum from the southern to northern limits of its native range (Darling et al., 2008). Although hybrids have not been directly quantified in the native range, gene flow likely has reduced differentiation among populations along the native cline, and adaptive differences and distinct population structure using genome-wide SNPs are observed at each extreme of the cline (Tepolt and Palumbi, 2015). This intraspecific split in the native range occurred more recently than the speciation event between C. maenas and its sister species C. aestuarii of five to eight million years ago based on COI sequence divergence calculated by Roman and Palumbi (2004). A similar time scale in the divergence of intraspecific ecotypes has recently been estimated in the European anchovy (E. encrasicolus) using genome-wide SNPs, where recent (post-glacial) secondary contact has occurred after long-term (c. 300 000 years) allopatric isolation (Le Moan et al., 2016). Our estimate here of divergence time for these green crab lineages further supports our previous conclusion of longstanding isolation and divergence (Jeffery et al., in press).
Interestingly, our estimations of migration events between green crab sampling sites showed substantial south to north gene flow to PLB, but both northwards and southwards gene flow towards KJI despite a southwest prevailing current consistent with previous work (Pringle et al., 2011). Given the distance and oceanographic conditions between these presumptive source populations and Placentia Bay, it is highly unlikely the migration events to this population represent natural dispersal and are most likely anthropogenic in origin (Blakeslee et al., 2010). The bidirectional migration observed at KJI is consistent with (natural) secondary contact between the established northern and southern populations in this region (Pringle et al., 2011; Darling et al., 2014). However, evidence also suggests the continued southwards displacement of northern haplotypes consistent with the prevailing currents in the region (Darling et al., 2014), indicative of a highly dynamic demographic system.
Implications of hybridization in green crabs
As previously discussed, hybridization, whether between separate species or among divergent populations within a species, can have a range of potentially negative or beneficial impacts. One obvious negative repercussion of hybridization to native ecosystems is if hybrids potentially have increased fitness relative to the parental populations or species, with environmental tolerances that encompass the range of the parents that allow them to rapidly expand their range (Burke and Arnold, 2001; Todesco et al., 2016). Within green crabs, the hybrid offspring detected in the present study are viable due to the presence of F2 and backcrossed hybrids despite their genomic divergence. From a management perspective, it may be imperative to treat green crabs as two ecotypes present in Atlantic Canada (Jeffery et al., in press), which likely have different thermal tolerances reflective of their native northern (Norway) and southern source populations (Roman, 2006; Tepolt and Somero, 2014). Secondary contact between these ecotypes appears to occur at the northern and southern limits of the southern and northern ecotypes, respectively in KJI and also in PLB. As 95% of the individual crabs we sampled at PLB were classified as hybrids, it is currently unknown whether the hybridization process has allowed this population to maintain itself in this location, possibly due to hybrid advantage or through repeated introduction of green crabs over time It is possible, though speculative, that hybrid individuals will have greater environmental tolerance relative to either parental ecotype, which may lead to their replacement over time (Gramlich et al., 2016; Todesco et al., 2016). Both hybrid advantage, and, the alternative, where hybrids simply show intermediate environmental tolerances to their parental linages, will require an examination of specific tolerances of temperature and growth rates as well as a re-evaluation of current mitigation strategies, which currently only consider green crab as a single ecotype in Canada (Therriault et al., 2008).
Summary
Here we identify the geographic extent to which hybridization between northern and southern green crab ecotypes occurs in eastern North America. We estimate that the European source populations of these ecotypes diverged ~470K years ago and have come into secondary contact in their invasive range in southern Nova Scotia. Both natural dispersal and anthropogenic transport appear to be driving hybridization, which is resulting in introgression and viable hybrid offspring between the lineages. The long-term effects of this hybridization are currently unknown, but may lead to greater thermal adaptation throughout their invasive range leading to continued range expansion, and even speciation or replacement of the parental ecotypes. Continued monitoring of green crab expansion will be necessary to determine when and where this species will ultimately be limited by the environment in eastern North America and what role hybridization among invasions will have.
Data Archiving
Raw RAD-seq reads are available in NCBI Sequence Read Archive BioProject PRNJA377723 (https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP102198). Aligned COI sequences are available from GenBank (Accession numbers KY800915-KY801176). Filtered genotype data for 9137 RAD-seq-derived SNPs, 13 microsatellites and simulated individuals for hybrid power analyses using both SNPs and microsatellites are available from the Dryad Digital Repository http://dx.doi.org/10.5061/dryad.p9b8k.
References
Abbott R, Albach D, Ansell S, Arntzen JW, Baird SJ, Bierne N et al. (2013). Hybridization and speciation. J Evol Biol 26: 229–246.
Anderson EC, Thompson EA . (2002). A model-based method for identifying species hybrids using multilocus genetic data. Genetics 160: 1217–1229.
Audet D, Davis DS, Miron G, Moriyasu M, Benhalima K, Campbell R . (2003). Geographical expansion of a nonindigenous crab, Carcinus maenas(L.), along the Nova Scotian shore into the southeastern Gulf of St. Lawrence, Canada. J Shellfish Res 22: 255–262.
Berrill M . (1982). The life cycle of the green crab Carcinus maenas at the northern end of its range. J Crust Biol 2: 31–39.
Besnier F, Glover KA . (2013). ParallelStructure: a R package to distribute parallel runs of the population genetics program structure on multi-core computers. PLoS ONE 8: e70651.
Blakeslee AMH, McKenzie CH, Darling JA, Byers JE, Pringle JM, Roman J . (2010). A hitchhiker’s guide to the Maritimes: anthropogenic transport facilitates long-distance dispersal of an invasive marine crab to Newfoundland. Divers Distrib 16: 879–891.
Bradbury IR, Bowman S, Borza T, Snelgrove PV, Hutchings JA, Berg PR et al. (2014). Long distance linkage disequilibrium and limited hybridization suggest cryptic speciation in Atlantic cod. PLoS ONE 9: e106380.
Buerkle C . (2005). Maximum‐likelihood estimation of a hybrid index based on molecular markers. Mol Ecol Notes 5: 684–687.
Burke JM, Arnold ML . (2001). Genetics and the fitness of hybrids. Ann Rev Genet 35: 31–52.
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013). Stacks: an analysis tool set for population genomics. Mol Ecol 22: 3124–3140.
Cristescu ME . (2015). Genetic reconstructions of invasion history. Mol Ecol 24: 2212–2225.
Darling JA . (2011). Interspecific hybridization and mitochondrial introgression in invasive Carcinus shore crabs. PLoS ONE 6: e17828.
Darling JA, Bagley MJ, Roman J, Tepolt CK, Geller JB . (2008). Genetic patterns across multiple introductions of the globally invasive crab genus Carcinus. Mol Ecol 17: 4992–5007.
Darling JA, Tsai YH, Blakeslee AM, Roman J . (2014). Are genes faster than crabs? Mitochondrial introgression exceeds larval dispersal during population expansion of the invasive crab Carcinus maenas. R Soc Open Sci 1: 140202.
Earl DA, vonHoldt BM . (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Res 4: 359–361.
Etter PD, Bassham S, Hohenlohe PA, Johnson EA, Cresko WA . (2011a) SNP discovery and genotyping for evolutionary genetics using RAD sequencing. In: Orgogozo V, Rockman MV (eds). Molecular Methods for Evolutionary Genetics, Humana Press: New York, NY, USA, pp 157–178.
Etter PD, Preston JL, Bassham S, Cresko WA, Johnson EA . (2011b). Local de novo assembly of RAD paired-end contigs using short sequencing reads. PLoS ONE 6: e18561.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Facon B, Jarne P, Pointier J, David P . (2005). Hybridization and invasiveness in the freshwater snail Melanoides tuberculata: hybrid vigour is more important than increase in genetic variance. J Evol Biol 18: 524–535.
Feder JL, Egan SP, Nosil P . (2012). The genomics of speciation-with-gene-flow. Trends Genet 28: 342–350.
Goudet J . (2005). Hierfstat, a package for R to compute and test hierarchical F‐statistics. Mol Ecol Notes 5: 184–186.
Gramlich S, Sagmeister P, Dullinger S, Hadacek F, Hörandl E . (2016). Evolution in situ: hybrid origin and establishment of willows (Salix L.) on alpine glacier forefields. Heredity 116: 531–541.
Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD . (2009). Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet 5: e1000695.
Harrison RG . (1993) Hybrid zones and the evolutionary process. Oxford University Press on Demand: New York, NY, USA.
Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, Cresko WA (2010). Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet 6: e1000862.
Hovick SM, Whitney KD . (2014). Hybridisation is associated with increased fecundity and size in invasive taxa: meta-analytic support for the hybridisation-invasion hypothesis. Ecol Lett 17: 1464–1477.
Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA . (2013). diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4: 782–788.
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I . (2015). Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15: 1179–1191.
Kovach AI, Walsh J, Ramsdell J, Kelley Thomas W . (2015). Development of diagnostic microsatellite markers from whole‐genome sequences of Ammodramus sparrows for assessing admixture in a hybrid zone. Ecol Evol 5: 2267–2283.
Le Moan A, Gagnaire PA, Bonhomme F . (2016). Parallel genetic divergence among coastal–marine ecotype pairs of European anchovy explained by differential introgression after secondary contact. Mol Ecol 25: 3187–3202.
Lee CE . (2002). Evolutionary genetics of invasive species. Trends Ecol Evol 17: 386–391.
Leigh JW, Bryant D . (2015). popart: full‐feature software for haplotype network construction. Methods Ecol Evol 6: 1110–1116.
Meirmans PG, Van Tienderen PH . (2004). GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4: 792–794.
Pickrell JK, Pritchard JK . (2012). Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet 8: e1002967.
Prentis PJ, Wilson JR, Dormontt EE, Richardson DM, Lowe AJ . (2008). Adaptive evolution in invasive species. Trends Plant Sci 13: 288–294.
Pringle JM, Blakeslee AM, Byers JE, Roman J . (2011). Asymmetric dispersal allows an upstream region to control population structure throughout a species’ range. Proc Nat Acad Sci USA 108: 15288–15293.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575.
Randi E . (2008). Detecting hybridization between wild species and their domesticated relatives. Mol Ecol 17: 285–293.
Reich D, Thangaraj K, Patterson N, Price AL, Singh L . (2009). Reconstructing Indian population history. Nature 461: 489–494.
Rius M, Darling JA . (2014). How important is intraspecific genetic admixture to the success of colonising populations? Trends Ecol Evol 29: 233–242.
Roman J . (2006). Diluting the founder effect: cryptic invasions expand a marine invader's range. Proc Biol Sci 273: 2453–2459.
Roman J, Palumbi SR . (2004). A global invader at home: population structure of the green crab, Carcinus maenas, in Europe. Mol Ecol 13: 2891–2898.
Sedghifar A, Brandvain Y, Ralph P . (2016). Beyond clines: lineages and haplotype blocks in hybrid zones. Mol Ecol 25: 2559–2576.
Seehausen O . (2013). Conditions when hybridization might predispose populations for adaptive radiation. J Exp Biol 26: 279–281.
Stanley RRE, Jeffery NW, Wringe BF, DiBacco C, Bradbury IR . (2017). GENEPOPEDIT: a simple and flexible tool for manipulating multilocus molecular data in R. Mol Ecol Res 17: 12–18.
Sundqvist L, Keenan K, Zackrisson M, Prodöhl P, Kleinhans D . (2016). Directional genetic differentiation and relative migration. Ecol Evol 6: 3461–3475.
Tepolt CK . (2014). Adaptation in marine invasion: a genetic perspective. Biol Invasions 17: 887–903.
Tepolt CK, Palumbi SR . (2015). Transcriptome sequencing reveals both neutral and adaptive genome dynamics in a marine invader. Mol Ecol 24: 4145–4158.
Tepolt CK, Somero GN . (2014). Master of all trades: thermal acclimation and adaptation of cardiac function in a broadly distributed marine invasive species, the European green crab, Carcinus maenas. J Expl Biol 217: 1129–1138.
Therriault TW, Herborg LM, Locke A, McKindsey CW . (2008). Risk assessment for European green crab (Carcinus maenas in Canadian waters. Canadian Science Advisory Secretariat, Fisheries and Oceans Canada: Victoria, BC, Canada.
Todesco M, Pascual MA, Owens GL, Ostevik KL, Moyers BT, Hübner S et al. (2016). Hybridization and extinction. Evol Appl 9: 892–908.
Twyford A, Ennos R . (2012). Next-generation hybridization and introgression. Heredity 108: 179–189.
Vähä JP, Primmer CR . (2006). Efficiency of model‐based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol Ecol 15: 63–72.
Viard F, David P, Darling JA . (2016). Marine invasions enter the genomic era: three lessons from the past, and the way forward. Curr Zool 62: 629–642.
Wringe BF, Stanley RRE, Jeffery NW, Anderson EC, Bradbury IR . (2017). Parallelnewhybrid: an R package for the parallelization of hybrid detection using NEWHYBRIDS. Mol Ecol Res 17: 91–95.
Xu S, Schaack S, Seyfert A, Choi E, Lynch M, Cristescu ME . (2012). High mutation rates in the mitochondrial genomes of Daphnia pulex. Mol Biol Evol 29: 763–769.
Zheng H, Xu F, Zhang G . (2011). Crosses between two subspecies of bay scallop Argopecten irradians and heterosis for yield traits at harvest. Aquac Res 42: 602–612.
Acknowledgements
We thank the staff at Fisheries and Oceans Canada for their assistance with sample collection, in particular, Jennifer FitzGerald, Cynthia McKenzie, Renée Bernier, Kyle Matheson, Philip Sargent, Erica Watson, Sophie Boudreau, Roland Hagan and April Blakeslee. We also thank the staff of the Aquatic Biotechnology Lab at the Bedford Institute of Oceanography for assistance with DNA extractions and RAD library preparation. Funding was provided by the Genomic Resource Development Initiative, Fisheries and Oceans Canada to IRB and CDB. This work was supported by a National Sciences and Engineering Research Council of Canada (NSERC) visiting fellowship to NWJ.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

Cite this article
Jeffery, N., DiBacco, C., Wringe, B. et al. Genomic evidence of hybridization between two independent invasions of European green crab (Carcinus maenas) in the Northwest Atlantic. Heredity 119, 154–165 (2017). https://doi.org/10.1038/hdy.2017.22
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2017.22
This article is cited by
-
Integrating seascape resistances and gene flow to produce area-based metrics of functional connectivity for marine conservation planning
Landscape Ecology (2023)
-
Challenges in eDNA detection of the invasive European green crab, Carcinus maenas
Biological Invasions (2022)
-
Natural hybridization among three Rhododendron species (Ericaceae) revealed by morphological and genomic evidence
BMC Plant Biology (2021)
-
Complex patterns of secondary spread without loss of genetic diversity in invasive populations of the Asian shore crab Hemigrapsus takanoi (Decapoda) along European coasts
Marine Biology (2020)
