
Abstract
Genomic selection (GS) is more efficient than traditional phenotype-based methods in hybrid breeding. The present study investigated the predictive ability of genomic best linear unbiased prediction models for rice hybrids based on the North Carolina mating design II, in which a total of 115 inbred rice lines were crossed with 5 male sterile lines. Using 8 traits of the 575 (115 × 5) hybrids from two environments, both univariate (UV) and multivariate (MV) prediction analyses, including additive and dominance effects, were performed. Using UV models, the prediction results of cross-validation indicated that including dominance effects could improve the predictive ability for some traits in rice hybrids. Additionally, we could take advantage of GS even for a low-heritability trait, such as grain yield per plant (GY), because a modest increase in the number of top selection could generate a higher, more stable mean phenotypic value for rice hybrids. Thus this strategy was used to select superior potential crosses between the 115 inbred lines and those between the 5 male sterile lines and other genotyped varieties. In our MV research, an MV model (MV-ADV) was developed utilizing a MV relationship matrix constructed with auxiliary variates. Based on joint analysis with multi-trait (MT) or with multi-environment, the prediction results confirmed the superiority of MV-ADV over an UV model, particularly in the MT scenario for a low-heritability target trait (such as GY), with highly correlated auxiliary traits. For a high-heritability trait (such as thousand-grain weight), MT prediction is unnecessary, and UV prediction is sufficient.
Similar content being viewed by others

Introduction
Genomic selection (GS) has been employed in both animal and plant breeding and potentially represents a more efficient method than traditional phenotype-based selection systems (Meuwissen et al., 2001; VanRaden et al., 2009). With the development of molecular biology, breeders can accurately understand the genetic structure of the population and thus greatly improve the efficiency of selection using whole-genome dense markers. Estimated breeding values based on the genotypes of individuals have been remarkably accurate (Resende et al., 2012; Lee et al., 2014). This accuracy has been verified in empirical studies in maize and rice (Technow et al., 2014; Onogi et al., 2015; Spindel et al., 2015). Previous studies have evaluated the efficacy of GS for breeding inbred lines of rice, and prediction accuracies ranged from 0.31 and 0.34 for grain yield and plant height to 0.63 for flowering time (Spindel et al., 2015). In predicting rice hybrid performance of an immortalized F2 population, the predictability ranges from 0.13 for yield to 0.68 for thousand-grain weight (TGW) (Xu et al., 2014).
The development of partial specific hybrid rice is a promising approach for exploiting the heterosis of rice (Adedze et al., 2016), and GS can be implemented to predict target traits of future hybrids with less hybrids identified in the field. In many cases, it is difficult to perform a complete diallel cross design because of the significant number of potential crosses or the use of some sterile lines. North Carolina mating design II (NC II) is a more feasible scheme in hybrid breeding including more parental species with a certain experimental scale. However, few GS studies in rice hybrids based on NC II have been undertaken. The present study aims to evaluate the predictive ability of GS for hybrid performance in rice based on the NC II scheme in which a total of 115 inbred rice lines were crossed with 5 male sterile lines and to demonstrate the application of predicting traits for potential crosses between the 115 inbred lines and those between the 5 male sterile lines and other genotyped varieties.
Non-additive genetic variation is typically ignored when genome-wide markers are used to perform genomic prediction of complex traits in plants or animals. However, dominance effects might have important roles in the genetic variation of complex traits (Wang et al., 2013). Incorporating dominance variances might help determine the total genetic contribution to a complex trait (Da et al., 2014). One of the goals of the present study was to investigate the effects of both additive and dominance variances in the genomic prediction of hybrid performance in rice based on NC II.
Most previous empirical studies with genome-wide prediction focused on within-environment predictions based on single-environment (SE) models, and these models typically target a single phenotypic trait. Modeling a single trait (ST) individually based on an SE disregards the fact that multiple traits (MTs) or phenotypes in multiple environments (MEs) are most likely associated, reflecting a shared biological basis (Scutari et al., 2014). Simultaneously, modeling multiple quantitative traits results in better predictive power than individually targeting traits (Henderson and Quaas, 1976; Hayashi and Iwata, 2013). In animal breeding, some traits (such as sex-linked traits or those expressed later in life) are difficult or expensive to measure. Some plant traits (such as root traits, stress resistance traits and grain yield per plot) also have similar characteristics. MT prediction with auxiliary traits that are easier or cheaper to record represents an effective strategy that avoids the use of genotypic information (Calus and Veerkamp, 2011). Additionally, recent studies have illustrated that the use of genetic and residual covariance across correlated environments might improve cross-environment accuracy in ME models (Burgueño et al., 2012; Guo et al., 2013; Heslot et al., 2014). In the present study, an efficient multivariate GS model (MV-ADV) was developed, utilizing a MV relationship matrix constructed with auxiliary variates (variates with the exception of the prediction target). An important objective of the present study was to investigate the performance of MV-ADV for predicting phenotypes of rice hybrids benefiting from joint analysis with auxiliary traits or with the phenotypes observed in other environments.
Materials and methods
Experimental data
The models were fitted to the rice data set from Wuhan University. A total of 115 inbred rice lines were crossed with 5 male sterile lines (including XinanS, Luohong3A, Y58S, Guangzhan63S and PA64S) via NC II. The 575 (115 × 5) crosses were evaluated in two environments in China, and two replicates were made in each environment.
Eight traits of rice were used in the present study, including grain yield per plant (GY), TGW, productive panicle number per plant (PN), plant height (PH), primary branch number (PB), secondary branch number (SB), grain number per panicle (GN) and panicle length (PL). Traits for the hybrid population and the inbred lines were collected during the 2013 rice growing season from replicated field trials on the experimental farms of Hubei Academy of Agricultural Sciences and Huazhong Agricultural University. The phenotypes fitting each model are the average performance of each line from two environments with the exception of the SE and ME analyses, where the phenotypes were averaged over two replications in a specific environment.
The inbred parents are genotyped using next-generation sequencing, and single-nucleotide polymorphisms (SNPs) were called using the Genome Analysis Toolkit (GATK 3.1). Let A1 and A2 represent two alleles at an SNP locus. The initial genetic values are subsequently given as −1, 0 and 1 for genotypes A1A1, A1A2 and A2A2, respectively. The SNPs were preprocessed after removing those with minor allele frequencies <5% and those with missing values in all the male sterile lines. The missing data in the remaining SNPs were replaced with the mean of the population at each locus. Finally, 3 299 150 high-density SNP markers were selected for analysis to infer the genotypes of the hybrids.
Additionally, using the 575 observed individuals as the training set, the performance of the potential crosses between the 5 male sterile lines and the 3023 rice varieties in the 3000-rice genomes project (Li et al., 2014) was predicted. The SNP genotyping data from both the 3000-rice genomes project and the present study were called against the Nipponbare reference genome. A total of 6 572 189 SNPs in the 3kRG filtered set (Alexandrov et al., 2015) were compared with the 3 299 150 SNPs, and subsequently the intersection, including 2 395 866 SNPs, was used to predict potential crosses between the 5 male sterile lines and the 3023 rice varieties.
Univariate models
Genomic best linear unbiased prediction (GBLUP) is an efficient method using whole-genome markers to predict genetic values and phenotypes of interest (Wang et al., 2015; Zhang et al., 2015). GBLUP includes a genomic relationship matrix in place of the traditional numerator relationship matrix A based on pedigree (VanRaden, 2008). The model can be described as:

where y is the vector of observations, b is the vector of non-genetic fixed effects, a is the vector of additive genetic effects, d is the vector of dominance effects and ɛ is the vector of random residuals. X is an incident matrix for the fixed effects and Z is an m × n incident matrix for random genetic effects (m is the number of phenotypic observations and n is the number of individuals). A univariate (UV) GBLUP model including only additive effects is called UV-A, whereas a UV GBLUP model including additive and dominance effects is called UV-AD.
It is assumed that  , where
, where  is the additive genetic variance;
is the additive genetic variance;  is the dominance variance;
is the dominance variance;  is the residual variance; Im is an m × m identity matrix; Ga and Gd are the additive and dominance genetic relationship matrices, respectively, constructed using information from genome-wide dense SNP markers.
is the residual variance; Im is an m × m identity matrix; Ga and Gd are the additive and dominance genetic relationship matrices, respectively, constructed using information from genome-wide dense SNP markers.
The additive genomic relationship matrix Ga can be obtained by

where Ma is the n × q matrix (q is the number of markers). The element of Ma for the ith individual at the jth marker is calculated as (Da et al., 2014; Nishio and Satoh, 2014):

where pj is the minor allele frequency at locus j. To avoid the inflation of variance components, a relationship matrix should be scaled to have average diagonal elements of 1 (Forni et al., 2011). In the present study, to obtain normalized matrices, ka was calculated as trace  .
.
The dominance genomic relationship matrix Gd can be obtained by

where Md is an n × q matrix and the element of Md for the ith individual at the jth marker is calculated as (Da et al., 2014; Nishio and Satoh, 2014):

The estimated additive effects (â) are obtained using the equation:

The inversion can be obtained because Im is full rank, and thus the sum within the square brackets can also be full rank. The estimated dominance effects (đ) are obtained using equations as indicated above. The proportions of the variances ( ) to the total variance (
) to the total variance ( ) were defined as
) were defined as  and
and  , respectively. The variance components and their proportions were estimated using restricted maximum likelihood (REML).
, respectively. The variance components and their proportions were estimated using restricted maximum likelihood (REML).
MV model
GS models were originally developed for an ST evaluated in an SE, and most analyses published thus far are based on UV models. Several studies have recently proposed using MV models for MEs or MTs (Jia and Jannink, 2012; Lopez-Cruz et al., 2015; Schulthess et al., 2016).
In the present study, a MV GS model MV-ADV was developed, which improved predictive performance through the use of a MV relationship matrix. For s traits or s environments, the MV-ADV model can be described as

where y is an m × 1 vector of the target variate and v is the vector of effects that can be reflected by the other variates. It is assumed that  ,
,  and
and  . Gv is the MV relationship matrix calculated as:
. Gv is the MV relationship matrix calculated as:

where Mv is the scaled MV matrix;  . yi is an n × 1 vector of observed phenotypic values of the ith trait or the ith environment, centred and standardized to unit variance. The phenotypes of the s-1 traits or environments were not the prediction targets and are called auxiliary variates.
. yi is an n × 1 vector of observed phenotypic values of the ith trait or the ith environment, centred and standardized to unit variance. The phenotypes of the s-1 traits or environments were not the prediction targets and are called auxiliary variates.  is the variance component of v, which can be estimated using REML.
is the variance component of v, which can be estimated using REML.
Cross-validation
Cross-validation was used to evaluate the performance of each previously described model. This statistical technique divides data into training and validation sets and uses the validation set to evaluate the predictive ability of the trained model. In each prediction, we randomly partitioned the data set into 5 parts of equal size and repeated the partitioning 20 times to obtain the average prediction results of 20 replicates from fivefold cross-validation. In the MV prediction, the cross-validation scheme mimicked a situation in which missing phenotypes of lines were predicted with auxiliary traits or the same trait recorded in a different environment. For example, in the two-environment scenario, the mean differences between two environments might produce inflated estimates of predictive ability, thus cross-validation with validation sets spanning different environments was prevented in the present study. In each cycle of prediction, the phenotypes of 115 hybrids in a certain environment were predicted based on the observed phenotypes of the other 460 hybrids in the same environment and 575 hybrids in the other environment.
Predictive ability was measured as the correlation coefficient between the observed phenotypes and estimated phenotypes of individuals in the validation set, adopted to evaluate the differences between models. Because the stochastic fivefold cross-validations were repeated 20 times for prediction, pairwise comparisons were used to test the differences in the predictive ability of different models. All analyses in the present study were performed using the R language, and the R program is provided in Supplementary Data set S1.
Inference of variance components
In the present study, the variance components and their proportions were estimated using REML. For the 20 stochastic fivefold cross-validations, the variance components should be efficiently estimated in each prediction iteration based on a specified training set. In the MV prediction, genetic or residual effects are often highly correlated between different traits or environments. Thus the estimation of the variance components might be close to singular, making the convergence slow. Therefore, the AI-REML algorithm (Lee and Van Der Werf, 2006) and the EM algorithm (Da et al., 2014) were implemented in the present study. The AI-REML algorithm has a rapid convergence rate (Ashida and Iwaisaki, 1999) and was the first choice in the analysis as implemented using R language. However, the AI-REML algorithm is not as robust as the EM algorithm and might be sensitive to the initial values of variance components (Wang et al., 2013). Thus the variance components were estimated using a combination of the AI-REML algorithm and the EM algorithm. Convergence was assumed when changes in the variance ratios between two successive rounds were <10−6.
Results
Prediction with UV models drawn from cross-validation
The prediction results of UV models drawn from cross-validation are summarized in Table 1. Using UV-A, GY showed the lowest predictive ability (0.3973); TGW and PH showed the highest predictive ability (0.8807 and 0.8603, respectively). This analysis shows that GS might be effective for all the traits of rice, particularly for TGW and PH. Because the individuals for prediction in the present study were hybrids based on NC II, not only the additive but also the dominance effects were considered in the UV models. The variance proportions estimated using REML showed that each trait is primarily controlled through additive variance. However, pairwise comparisons illustrated that predictive ability of UV-AD was significantly higher than that of UV-A for PH, PB, SB and GN, showing that, for these traits, including dominance effects could improve the predictive ability. For GY, TGW, PN and PL, there were no significant differences between the predictive ability of UV-A and UV-AD, showing that it is safe to include dominance variances in the model, even when a trait is only controlled through additive variance. In the MV model, to optimize the prediction and facilitate the comparison, both additive and dominance effects were included. Additionally, prediction for the 115 inbred rice lines was conducted using UV-A in the present study. Reflecting a smaller training set size or a different relationship structure, the prediction results for the inbred lines were substantially lower than those for the hybrids with the exception of trait PN (Figure 1).

Average predictive ability of UV-A for the 115 inbred lines and UV-AD for the 575 hybrids.
Predictive ability was adopted to evaluate the above prediction results. However, breeders are concerned with the phenotypic values of the selected crosses. In each cycle of the cross-validation, we sorted the predicted phenotypic values in descending order and selected different number of top crosses to observe the benefits of prediction. Figure 2 shows the mean percentage increase in the true phenotypic values of top crosses relative to the mean of each validation set plotted against the number of top crosses being selected for each trait. With 100 predictions (from 20 stochastic fivefold cross-validations) performed and top 10 crosses being selected for GY, although the mean percentage increase was 16.1%, the s.d. of the percentage increase reached 7.8%. For a high-heritability trait, such as TGW or PH, the s.d. was consistently at a low level, though the mean percentage increase was not as high as we expected. Obviously, the trouble of predicting a low-heritability trait, such as GY, is not the small mean of phenotypic values but the significant s.d.. With top 30 crosses being selected, the percentage increase was 10.6% and the s.d. was reduced to 3.1%, showing that a modest increase in the number of top selection could generate a stable higher mean phenotypic value for rice hybrids. In a word, it is exciting that we can take advantage of GS even for a trait with low predictive ability.

Mean percentage increase in the true phenotypic values of top crosses relative to the mean of each validation set plotted against the number of top selection. The upper red dotted curves define the mean percentage increase in the true phenotypic values of top crosses plus the s.d. of 100 predictions (drawn from 20 stochastic fivefold cross-validations). The lower red dotted curves define the mean percentage increase in the true phenotypic values of top crosses minus the s.d., GY, TGW, PN and PL were predicted using UV-A; PH, PB, SB and GN were predicted using UV-AD.
Prediction for potential crosses
With the 575 observed individuals as a training set, performance of the potential crosses between the 115 inbred lines was predicted. Using UV-A, the predicted phenotypic values of GY for the 6555 crosses are given in Supplementary Data set S2. The predicted GY were sorted in descending order; thus the top 100 crosses were selected. The mean predicted GY of the top 100 crosses is 51.78±1.38, which is much higher than the average predicted GY of the entire hybrid population (38.94). Furthermore, we noticed that 42 of the top 100 shared the same parent Huangxiuzhan and 39 of the top 100 were the progeny of R238, indicating that Huangxiuzhan and R238 are more likely to be utilized to produce high-performing hybrids for GY.
Additionally, with the 575 hybrids as a training set, performance of the potential crosses between the 5 male sterile lines and the 3023 rice varieties in the 3000-rice genomes project was predicted. Using UV-A, the predicted phenotypic values of GY for the 15 115 crosses are given in Supplementary Data set S3. The top 100 crosses for GY were selected, with 93 of them sharing the same parent Guangzhan63S and 7 of them being the progeny of PA64S. The mean predicted GY of the top 100 crosses is 44.43±0.52. The average predicted GY of the entire hybrid population is 38.50. Breeders can actually produce these top crosses based on the results of the present study.
Prediction with MTs
An MT genomic model might have higher prediction accuracy than an ST genomic model. Regarding the hybrid performance, the prediction results of MV-ADV with two traits are illustrated in Table 2. As shown, the performance of MV-ADV was significantly superior to UV-AD. Trait GY showed the most significant improvement in MT prediction, particularly with PN as an auxiliary trait, showing that making use of observed traits related to the target trait can help enhance predictive ability in MT prediction. PN, GN and SB also showed an improvement in the MT prediction. The predictive ability of TGW hardly increased, no matter which trait was used as an auxiliary trait. The above results demonstrated the benefit of MT prediction for traits with low heritability, such as GY. For a high-heritability trait, such as TGW, MT prediction is unnecessary, and UV prediction is a good choice.
In MV-ADV, the MV effect v is an important component deserving attention. The percentage increase in the predictive ability of MV-ADV relative to UV-AD plotted against the variance proportion of v to the total variance ( ) estimated using REML is presented in Figure 3. In most cases, the percentage increase in predictive ability is positively correlated with
) estimated using REML is presented in Figure 3. In most cases, the percentage increase in predictive ability is positively correlated with  , showing that the MV effect v is the main contributor to the gains with MT over ST.
, showing that the MV effect v is the main contributor to the gains with MT over ST.

Percentage increase in the predictive ability of MV-ADV relative to UV-AD plotted against  estimated using REML.
estimated using REML.
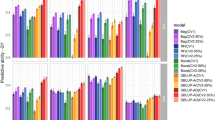
In the present MT study, not only two traits were considered at a time but also all of the eight traits were included. Figure 4 describes the average prediction results of UV-AD with an ST, MV-ADV with two traits and MV-ADV with eight traits for the hybrids. GY and PN showed the most significant increases in MT prediction with eight traits, while the predictive ability of TGW and PH were slightly improved, showing again that the MT strategy was more suitable for a trait with low heritability. The average predictive ability of MV-ADV with two traits was 6.4% higher than that of UV-AD and the gains of MV-ADV with eight traits over UV-AD was 26.7%. As shown, MV-ADV with eight traits yielded substantial increases in predictive ability relative to MV-ADV with two traits and UV-AD with an ST, demonstrating that a joint analysis with more auxiliary traits might greatly improve the predictive ability.

Average predictive ability of UV-AD with a single trait, MV-ADV with two traits and MV-ADV with eight traits for the hybrids.
Prediction with MEs
To minimize the effects of error on prediction, the phenotypic values of each trait used in the aforementioned analysis are the average of each hybrid from two environments. Based on UV-AD, a comparison of the prediction results for the hybrids using phenotypic values from an SE and the average of two environments are shown in Table 3. Obviously, using average phenotypes from MEs might improve the predictive ability.
Furthermore, accuracy improvements in the ME scenario were evaluated in the present study. A comparison between the predictive ability of the SE versus ME model is shown in Table 4, where UV-AD represents the average of two SE prediction results, and the predictive ability of MV-ADV is the average of two ME prediction results (with each environment as the prediction target). Clearly, the performance of MV-ADV was better than that of UV-AD. In these ME analyses, the highest  was produced for TGW (0.2330), followed by that for PB (0.1252), resulting in positive gains of MV-ADV over UV-AD. The smallest
was produced for TGW (0.2330), followed by that for PB (0.1252), resulting in positive gains of MV-ADV over UV-AD. The smallest  (0.0076) was detected in the ME analysis for PN, and thus the ME prediction underperformed the SE prediction, showing that the MV effect v has an important role in the ME prediction.
(0.0076) was detected in the ME analysis for PN, and thus the ME prediction underperformed the SE prediction, showing that the MV effect v has an important role in the ME prediction.
Discussion
Effect of dominance variance components on prediction
Previous studies have shown that dominance is the main contributor to heterosis for height in rice (Shen et al., 2014). Huang et al. (2015) also found that accumulation of alleles with positive dominance is an important contributor to the heterotic phenomena in rice hybrids. Thus detecting some dominance effects would be expected in the rice hybrids based on NC II. However, in the present study, for most of the eight traits, little dominance variances were detected, showing that the traits are primarily controlled through additive variance. Previous studies have shown that fitting both additive and dominance effects of marker genotypes provided either similar or higher accuracy depending on the value of dominance variance (Sun et al., 2014). For some traits with bigger dominance variances, we observed the effects of variance components on predictive ability. For trait PB in hybrids, Table 1 shows that UV-AD outperforms UV-A the most in predictive ability; accordingly, the variance accounted for by dominance is the most significant (7.89%), illustrating that adding a substantial dominance component estimated using REML can substantially contribute to hybrid prediction.
Effect of marker density on prediction
The impacts of marker density were also investigated using the UV-AD model. Four SNP densities were compared using UV-AD for the rice hybrids (Supplementary Table S1). The edited set of 3 299 150 SNPs with >5% minor allele frequency (designated as 3300K) was compared with subsets of those SNPs: 1 609 838 (1610K), 470 688 (470K), and 95 994 (96K). The 1610K and 470K subsets were obtained by removing the SNPs with the missing rates >10% and >2%, respectively; the 96K subset was obtained by only retaining the SNPs without any missing values.
The average predictive ability slightly increased as the density of SNP markers increased from 96K to 3300K, showing that large SNP sets with missing values could also undergo improved prediction accuracy to a certain degree. Lumping consecutive SNP sites with the same genotype from the 3300K set was also tried in the present study, but the prediction result was almost the same as that with the 3300K. Previous studies have shown that more markers can increase the accuracy of GS by providing SNPs located closer to the causative genes (VanRaden et al., 2009). In the current study, gains in 3300K might be attributed to more information provided by ultra-high-density SNPs. Therefore, the 3300K SNP set was chosen to construct genetic relationship matrices in the present study.
Value of the NC II scheme for predicting some potential crosses
In the present study, with phenotypic values of rice hybrids base on NC II and high-density SNP markers, cross-validation was used to evaluate the accuracy of each previously described prediction. Application of GS for predicting rice hybrids derived from recombinant inbred lines has been reported previously (Xu et al., 2014). The 575 hybrids can certainly be used as a training sample to predict traits of some important potential crosses. First, performance of the crosses between the 115 inbred lines can be predicted as described previously. Second, there are many varieties of rice already genotyped, such as the 3023 rice varieties in the 3000-rice genomes project and the 1495 diverse varieties of hybrid rice (Huang et al., 2015). Theoretically, the hybrids for prediction do not have to share common parents with the hybrids in the training set. Nevertheless, it is notable that these experimental data are based on NC II and the five male sterile lines have an important role in the performance of the hybrids. Using the 575 hybrids as the training set, to ensure the accuracy of prediction, prediction of some potential crosses between the 5 male sterile lines and several other varieties is recommended.
High-quality shared marker is an important factor for genomic prediction. As discussed previously, the accuracy of GS is expected to be increased with more SNPs located closer to the causative genes. From the 3000-rice genomes project, when 996 009 SNPs in the 3kRG Core SNP set (Alexandrov et al., 2015) was compared with the 3 299 150 SNPs, the intersection included only 155 833 SNPs, leading to a great loss of information. Using 6 572 189 SNPs in the 3kRG filtered set (Alexandrov et al., 2015), the intersection including 2 395 866 SNPs was selected, and most of the marker information could be kept. Obviously, to find more common markers, using high-density SNPs of the prediction target was necessary. Additionally, 42 varieties in the 3000-rice genomes project were found to have the same names with some varieties in the 115 inbred lines, and consistency of their SNPs was checked. Ignoring heterotic and missing markers, the average consistency rate of the SNPs in the homonymous varieties was 0.8604±0.1520. Notably, the consistency rates of the 15 varieties were >0.99 and those of 7 varieties were <0.70. We presumed that some inconsistency reflected the practical differences between homonymous varieties provided by different units. Taking these differences into account, the 42 homonymous varieties were not excluded from the 3023 varieties for prediction. Thus the quality of the SNP data was reliable, and the 575 observed hybrids can be used as a training set for predicting some potential crosses in rice.
Effect of the MV model on MT prediction
The joint analysis of MTs has been shown to help improve the selection of some primary traits with low heritabilities or those that are difficult to measure (Alimi et al., 2013). Recent studies have illustrated that MT models might improve the accuracy of prediction. MT models are most advantageous when the traits analyzed are highly correlated (Piepho et al., 2008). An MT model is expected to increase the accuracy of prediction by incorporating information from genetically correlated traits. The phenotypic correlation between traits contains both genetic and error correlations. Therefore, the performance of MT models is associated with the correlation coefficient between the phenotypic values of MTs (Supplementary Table S2). The highest correlation coefficient was produced between SB and GN followed by that between GY and PN. As expected, joint analyses with SB and GN or GY and PN produced notable improvements in prediction accuracy.
Although genetic correlation was shown to be a key factor in determining the MT advantage over ST (Jia and Jannink, 2012), results in the present study show that there are other important factors affecting the advantage. In the MT prediction with two traits, the improvement of joint analyses with SB and GN was not on the top of list, which was much smaller than that with GY and PN. On average, GY showed the most significant improvement, followed by PN, GN and SB, indicating that low-heritability traits might be better predicted using an MT model. Furthermore, MT and ST analyses did not produce significantly different prediction accuracies for a trait with high heritability, such as TGW or PH. In a word, MV-ADV is best suited for a target trait with low heritability or the joint analysis of highly correlated traits.
The MV relationship matrix of MV-ADV is constructed with the phenotypic values, which reflects not only the shared biological basis but also the shared environmental effects in MT prediction. The highest phenotypic correlation coefficient was produced between GN and SB (Supplementary Table S2), and thus the most significant variance proportion of  (0.4891) was achieved in predicting GN with SB, followed by predicting SB with GN (0.3984). However, joint analyses of GN and SB did not produce high-level increase in the predictive ability of MV-ADV relative to UV-AD. We presume that the lack of benefits reflected the higher heritability of GN and SB. The genetic effects in UV-AD reflect the phenotypic variation, leading to the less contribution of the MV effect v to the predictive ability.
(0.4891) was achieved in predicting GN with SB, followed by predicting SB with GN (0.3984). However, joint analyses of GN and SB did not produce high-level increase in the predictive ability of MV-ADV relative to UV-AD. We presume that the lack of benefits reflected the higher heritability of GN and SB. The genetic effects in UV-AD reflect the phenotypic variation, leading to the less contribution of the MV effect v to the predictive ability.
Previous studies have shown that the contributions of auxiliary traits added to the model would tend asymptotically toward zero; therefore, MT prediction with two traits should be sufficient to improve the prediction accuracy for a particular trait (Schulthess et al., 2016). However, in the MV analyses, predictive ability of GY with eight traits using MV-ADV was 0.8794, showing notable increase not only relative to the average predictive ability with two traits (0.5217) but also relative to the highest predictive ability with two traits (0.7590). The scenario of predicting PN was similar to that of predicting GY, showing that a joint analysis with more auxiliary traits might greatly improve the predictive ability. In plant breeding, traits being sex-linked or expressed later in life are not as widespread as those in animals. However, some plant traits are difficult or expensive to measure, such as root traits, stress resistance traits and grain yield per plot, and thus the application of MT prediction borrowing phenotypic values of auxiliary traits from the target population is promising.
Moreover, traditional MV methods are known for using genetic and residual covariance across correlated traits to obtain sizable gains in prediction accuracy (Scutari et al., 2014; Lopez-Cruz et al., 2015). These methods are more time-consuming than UV methods, and this limitation is expected to become serious when the number of phenotypic records or variates increases (Schulthess et al., 2016). In the present study, the dimensions of MV-ADV are the same as those of UV models (n × n), leading to high efficiency particularly in REML.
Effect of the MV model on ME prediction
In our ME analyses, the MV model for ME prediction includes all data to estimate marker effects, and borrowing information across environments is permitted. For most of the eight traits, the prediction can benefit from records of the hybrids collected in other environments, which is consistent with previous across-environment work (Jarquín et al., 2014). However, it is notable that, in the ME analysis for PN and PH, the ME prediction underperformed the SE prediction, showing that environmental difference might negatively affect the ME prediction in some cases. Additionally, as shown in Tables 3 and 4, MV-ADV underperformed the prediction for average phenotypic values of two environments, showing that the latter is a feasible method to eliminate the effects of error on prediction. If the goal is to obtain a maximal amount of genetic information regarding the target plant, then replicated field trials on several farms in different environments are necessary, and using average phenotypic values of different environments is simple and effective. However, if the research focuses on plant performance in a specified environment, then trials in other environments are valuable, and the MV-ADV model is recommended.
Conclusions
The prediction results of the rice hybrid performance based on NC II demonstrated that we could take advantage of GS, even for a low-heritability trait (such as GY), because a modest increase in the number of top selection could generate a stable higher mean phenotypic value for rice hybrids. Thus the strategy was used to pick out superior potential crosses between the 115 inbred lines and those between the 5 male sterile lines and some other genotyped varieties. Additionally, in the present study, we developed an MV-ADV model for predicting unobserved phenotypes of hybrids benefiting from joint analysis with auxiliary traits or with the phenotypes observed in other environments. In both MT and ME prediction, the results confirmed the superiority of MV-ADV over UV-AD, particularly in the MT scenario for a low-heritability target trait (such as GY) with highly correlated auxiliary traits. For a high-heritability trait (such as TGW), MT prediction is unnecessary, and UV prediction is sufficient. We expect that the outcomes of the present study will contribute to the genomic prediction of rice hybrid performance.
Data archiving
Phenotype and genotype data are available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.kg26j.
References
Adedze YMN, He WC, Samoura AD, Huang F, Tondi YN, Efisue A et al. (2016). Genomic composition and yield heterosis of the partial inter-specific hybrid rice between Oryza sativa L. and Oryza glaberrima Steud. J Agric Sci 154: 367–382.
Alexandrov N, Tai S, Wang W, Mansueto L, Palis K, Fuentes RR et al. (2015). SNP-Seek database of SNPs derived from 3000 rice genomes. Nucleic Acids Res 43: D1023–D1027.
Alimi NA, MCAM Bink, Dieleman JA, Magán JJ, Wubs AM, Palloix A et al. (2013). Multi-trait and multi-environment QTL analyses of yield and a set of physiological traits in pepper. Theor Appl Genet 126: 2597–2625.
Ashida I, Iwaisaki H . (1999). An expression for average information matrix for a mixed linear multi-component of variance model and REML iteration equations. Anim Sci J 70: 282–289.
Burgueño J, de los Campos G, Weigel K, Crossa J . (2012). Genomic prediction of breeding values when modeling genotype × environment interaction using pedigree and dense molecular markers. Crop Sci 52: 707–719.
Calus MP, Veerkamp RF . (2011). Accuracy of multi-trait genomic selection using different methods. Genet Sel Evol 43: 1–14.
Da Y, Wang C, Wang S, Hu G . (2014). Mixed model methods for genomic prediction and variance component estimation of additive and dominance effects using SNP markers. PLos One 9: e87666.
Forni S, Aguilar I, Misztal I . (2011). Different genomic relationship matrices for single-step analysis using phenotypic, pedigree and genomic information. Genet Sel Evol 43: 43–53.
Guo Z, Tucker DM, Wang D, Basten CJ, Ersoz E, Briggs WH et al. (2013). Accuracy of across-environment genome-wide prediction in maize nested association mapping populations. G3 (Bethesda) 3: 263–272.
Hayashi T, Iwata H . (2013). A Bayesian method and its variational approximation for prediction of genomic breeding values in multiple traits. BMC Bioinformatics 14: 34.
Henderson C, Quaas R . (1976). Multiple trait evaluation using relatives' records. J Anim Sci 43: 1188–1197.
Heslot N, Akdemir D, Sorrells ME, Jannink J-L . (2014). Integrating environmental covariates and crop modeling into the genomic selection framework to predict genotype by environment interactions. Theor Appl Genet 127: 463–480.
Huang X, Yang S, Gong J, Zhao Y, Feng Q, Gong H et al. (2015). Genomic analysis of hybrid rice varieties reveals numerous superior alleles that contribute to heterosis. Nat Commun 6: 6258.
Jarquín D, Crossa J, Lacaze X, Cheyron PD, Daucourt J, Lorgeou J et al. (2014). A reaction norm model for genomic selection using high-dimensional genomic and environmental data. Theor Appl Genet 127: 595–607.
Jia Y, Jannink J-L . (2012). Multiple-trait genomic selection methods increase genetic value prediction accuracy. Genetics 192: 1513–1522.
Lee SH, Van Der Werf JH . (2006). An efficient variance component approach implementing an average information REML suitable for combined LD and linkage mapping with a general complex pedigree. Genet Sel Evol 38: 1–19.
Lee Y-S, Kim H-J, Cho S, Kim H . (2014). The usage of an SNP-SNP relationship matrix for best linear unbiased prediction (BLUP) analysis using a community-based cohort study. Genomics Inform 12: 254–260.
Li J-Y, Wang J, Zeigler RS . (2014). The 3,000 rice genomes project: new opportunities and challenges for future rice research. Gigascience 3: 8.
Lopez-Cruz M, Crossa J, Bonnett D, Dreisigacker S, Poland J, Jannink J-L et al. (2015). Increased prediction accuracy in wheat breeding trials using a marker × environment interaction genomic selection model. G3 (Bethesda) 5: 569–582.
Meuwissen TH, Hayes BJ, Goddard ME . (2001). Prediction of total genetic value using genome-wide dense marker maps. Genetics 157: 1819–1829.
Nishio M, Satoh M . (2014). Including dominance effects in the genomic BLUP method for genomic evaluation. PLoS One 9: e85792.
Onogi A, Ideta O, Inoshita Y, Ebana K, Yoshioka T, Yamasaki M et al. (2015). Exploring the areas of applicability of whole-genome prediction methods for Asian rice (Oryza sativa L.). Theor Appl Genet 128: 41–53.
Piepho HP, Möhring J, Melchinger AE, Büchse A . (2008). BLUP for phenotypic selection in plant breeding and variety testing. Euphytica 161: 209–228.
Resende MF, Muñoz P, Resende MD, Garrick DJ, Fernando RL, Davis JM et al. (2012). Accuracy of genomic selection methods in a standard data set of loblolly pine (Pinus taeda L.). Genetics 190: 1503–1510.
Schulthess AW, Wang Y, Miedaner T, Wilde P, Reif JC, Zhao Y . (2016). Multiple-trait- and selection indices-genomic predictions for grain yield and protein content in rye for feeding purposes. Theor Appl Genet 129: 273–287.
Scutari M, Howell P, Balding DJ, Mackay I . (2014). Multiple quantitative trait analysis using Bayesian networks. Genetics 198: 129–137.
Shen G, Zhan W, Chen H, Xing Y . (2014). Dominance and epistasis are the main contributors to heterosis for plant height in rice. Plant Sci 215: 11–18.
Spindel J, Begum H, Akdemir D, Virk P, Collard B, Redona E et al. (2015). Genomic selection and association mapping in rice (Oryza sativa: effect of trait genetic architecture, training population composition, marker number and statistical model on accuracy of rice genomic selection in elite, tropical rice breeding lines. PLoS Genet 11: e1004982.
Sun C, Vanraden PM, Cole JB, O'Connell J . (2014). Increasing predictive ability using dominance in genomic selection. Proceedings, 10th World Congress of Genetics Applied to Livestock Production. Vancouver, Canada.
Technow F, Schrag TA, Schipprack W, Bauer E, Simianer H, Melchinger AE . (2014). Genome properties and prospects of genomic prediction of hybrid performance in a breeding program of maize. Genetics 197: 1343–U1469.
VanRaden P . (2008). Efficient methods to compute genomic predictions. J Dairy Sci 91: 4414–4423.
VanRaden P, Van Tassell C, Wiggans G, Sonstegard T, Schnabel R, Taylor J et al. (2009). Invited review: reliability of genomic predictions for North American Holstein bulls. J Dairy Sci 92: 16–24.
Wang C, Prakapenka D, Wang S, Runesha H, Da Y . (2013). GVCBLUP: a computer package for genomic prediction and variance component estimation of additive and dominance effects. BMC Bioinformatics 15: 270.
Wang X, Yang Z, Xu C . (2015). A comparison of genomic selection methods for breeding value prediction. Sci Bull 60: 925–935.
Xu S, Zhu D, Zhang Q . (2014). Predicting hybrid performance in rice using genomic best linear unbiased prediction. Proc Natl Acad Sci USA 111: 12456–12461.
Zhang Z, Erbe M, He J, Ober U, Gao N, Zhang H et al. (2015). Accuracy of whole-genome prediction using a genetic architecture-enhanced variance-covariance matrix. G3 (Bethesda) 5: 615–627.
Acknowledgements
This work was supported by grants from the National Key Technology Research and Development Program of MOST (2016YFD0100303), the Priority Academic Program Development of Jiangsu Higher Education Institutions, the National Natural Science Foundations (91535103, 31391632 and 31200943), the National High-tech R&D Program (863 Program) (2014AA10A601-5), the Natural Science Foundations of Jiangsu Province (BK2012261), the Natural Science Foundation of the Jiangsu Higher Education Institutions (14KJA210005), the Postgraduate Research and Innovation Project in Jiangsu Province (KYLX151368) and the Innovative Research Team of Universities in Jiangsu Province.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Rights and permissions
About this article

Cite this article
Wang, X., Li, L., Yang, Z. et al. Predicting rice hybrid performance using univariate and multivariate GBLUP models based on North Carolina mating design II. Heredity 118, 302–310 (2017). https://doi.org/10.1038/hdy.2016.87
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2016.87
This article is cited by
-
Improving hybrid rice breeding programs via stochastic simulations: number of parents, number of hybrids, tester update, and genomic prediction of hybrid performance
Theoretical and Applied Genetics (2024)
-
Review of applications of artificial intelligence (AI) methods in crop research
Journal of Applied Genetics (2024)
-
Sparse Phenotyping and Haplotype-Based Models for Genomic Prediction in Rice
Rice (2023)
-
Optimization of Multi-Generation Multi-location Genomic Prediction Models for Recurrent Genomic Selection in an Upland Rice Population
Rice (2023)
-
A statistical package for evaluation of hybrid performance in plant breeding via genomic selection
Scientific Reports (2023)
