
Abstract
As deforestation and urbanization continue at rapid rates in tropical regions, urban forest patches are essential repositories of biodiversity. However, almost nothing is known about gene flow of forest-dependent tree species in urban landscapes. In this study, we investigated gene flow in the insect-pollinated, wind-dispersed tropical tree Koompassia malaccensis in and among three remnant forest patches in the urbanized landscape of Singapore. We genotyped the vast majority of adults (N=179) and a large number of recruits (N=2103) with 8 highly polymorphic microsatellite markers. Spatial genetic structure of the recruit and adult cohorts was significant, showing routine gene dispersal distances of ~100–400 m. Parentage analysis showed that 97% of recruits were within 100 m of their mother tree, and a high frequency of relatively short-distance pollen dispersal (median ~143–187 m). Despite routine seed and pollen dispersal distances of within a few hundred meters, interpatch gene flow occurred between all patches and was dominated by pollen movement: parentage analysis showed 76 pollen versus 2 seed interpatch dispersal events, and the seedling neighborhood model estimated ~1–6% seed immigration and ~21–46% pollen immigration rates, depending on patch. In addition, the smallest patch (containing five adult K. malaccensis trees) was entirely surrounded by >2.5 km of ‘impervious’ substrate, yet had the highest proportional pollen and seed immigration estimates of any patch. Hence, contrary to our hypothesis, insect-mediated gene flow persisted across an urban landscape, and several of our results also parallel key findings from insect-pollinated canopy trees sampled in mixed agricultural–forest landscapes.
Similar content being viewed by others
Introduction
Across the tropics, primary forests are being degraded and fragmented in rural, suburban and urban settings (DeFries et al., 2010; Gao and Yu, 2014). Urban population growth outpaces rural growth, and hence increasingly more areas of tropical forests will be reduced to urban forest patches. These patches may be of high conservation value and retain important biodiversity today (Alvey, 2006; Seamon et al., 2006), but the long-term persistence of biodiversity in increasingly urbanized landscapes remains a conservation concern.
Studies of genetic structuring and connectivity of plants in partially deforested landscapes have largely focused on mixed agricultural–forest and pastoral systems, secondary forest or recently logged forest (reviewed in Aguilar et al., 2008; Eckert et al., 2010). These studies have shown that fragmentation can disrupt relationships between plants and their biotic pollen and/or seed dispersers via changes to plant population demography as well as animal abundance, diversity and behavior, with follow-on effects to plant mating systems and progeny (reviewed in Ghazoul, 2005; Aguilar et al., 2008; Eckert et al., 2010).
Studies on gene flow of insect-pollinated trees in agricultural and modified forest landscapes have documented several trends. First, many bee species are capable of traversing long distances across this landscape (up to 14 km), suggesting that these matrices may not be a barrier to gene flow (reviewed in Dick et al., 2008). Second, there can often be a higher magnitude of long-distance gene flow involving isolated trees or isolated stands compared with more continuous forest or among larger patches (see, for example, Aldrich and Hamrick, 1998; Lander et al., 2010; Fuchs and Hamrick, 2011; Ismail et al., 2012; Rymer et al., 2013; Tambarussi et al., 2015; Guidugli et al., 2016). Third, individual trees can be disproportionally influential to interpatch connectivity (see, for example, Aldrich and Hamrick, 1998; Lander et al., 2010; de Moraes and Sebbenn, 2011; Fuchs and Hamrick, 2011; Ismail et al., 2012). Finally, trees in modified habitats (mostly agricultural) frequently experience increased rates of self-fertilization (‘selfing’) (reviewed in Ferreira et al., 2013).
There has been almost no research on gene flow among tree populations in urban landscapes, despite the important conservation implications. The few existing studies (Wang et al., 2010; Sebbenn et al., 2011; Nagamitsu et al., 2014) have examined wind-pollinated tree species or trees in single isolated forest patches, limiting wider application to urban forest fragment matrices, especially in the tropics where plants are almost exclusively animal pollinated (Ollerton et al., 2011). In contrast to agricultural or forested landscapes, the urban matrix often consists of dense built-up areas, suburban housing, industrial buildings, managed parks and tiny remnant forest fragments. The presence of ‘impervious’ habitat—for example, highways, parking lots, buildings—has been shown to act as a stronger filter or barrier to the movement of small, mobile animals between suitable habitats (for example, bees, Davis et al., 2010; Jha and Kremen, 2013; beetles, Keller and Largiadèr, 2003; frogs, Hitchings and Beebee, 1997; salamanders, Noël et al., 2007; birds and lizards, Delaney et al., 2010; and mice, Munshi-South and Kharchenko, 2010). Hence, as opposed to an agricultural or modified forest matrix, an ‘impervious’ urban matrix could reduce animal-mediated gene flow among forest patches. This would result in long-term genetic consequences including accelerated genetic erosion and genetic drift, increased inbreeding and biparental inbreeding and reduced pollen diversity, leading to a loss of fitness in progeny (see, for example, Breed et al., 2012; Rymer et al., 2013).
In a previous paper, Noreen and Webb (2013) showed that despite a dramatic reduction of habitat area in Singapore in the past ~150 years (to <0.2% of the original primary forest area; Corlett, 1992), genetic diversity was still high in adult, juvenile and seedling cohorts of the large canopy tree species Koompassia malaccensis Maingay ex Benth. (Fabaceae). There was no evidence of a population bottleneck, suggesting that overall the Singapore population of K. malaccensis appeared to have resisted genetic erosion (Noreen and Webb, 2013). However, understanding connectivity between patches is necessary to obtain a more complete picture of the dynamics of gene flow and of the possible fate of remnant rainforest populations in urban landscapes; genetic analyses provide information on the reproductive patterns of the adults and the final outcome of pollen and seed dispersal patterns as shown by population genetics of established recruits.
In this study, we investigated patterns of gene flow in the insect-pollinated, wind-dispersed and long-lived tropical tree K. malaccensis in remnant primary forest fragments in the tropical urbanized landscape of Singapore. We tested the hypothesis that insect-mediated gene flow among urban forest patches would be restricted. To test this hypothesis, we conducted a comprehensive parentage analysis and estimated immigration rates for each patch using a spatially explicit seedling neighborhood model. We also estimated genetic indices, selfing rates and the spatial genetic structure (SGS) of the recruit cohorts in each patch as well as the adults.
Materials and methods
Study species
K. malaccensis is a widespread, commercially valuable Southeast Asian tree species. It is long-lived and has a density of ~1 individual per hectare in primary forest. In Singapore, extensive clearing for agriculture (90% cleared by 1900) has relegated the remaining adults to the few remaining primary forest fragments, along with scattered remnant individuals in secondary forest (Corlett, 1992; Yee et al., 2011). The average diameter growth rate for K. malaccensis adults (stem diameter of >30 cm), averaged ~0.59 cm per year from 1993 to 2008 in Bukit Timah Nature Reserve (N=15 adults; SKY Lum, unpublished data), suggesting that most present-day adult trees are well over a century old and hence likely pre-date much of the deforestation in Singapore.
K. malaccensis has a mixed mating system: it is a preferential outcrosser but is able to self-fertilize (Lee et al., 2011; Noreen and Webb, 2013). The flowers are small (~3 mm diameter), white and insect pollinated, and the fruits are flattened, lightweight single-seeded pods ~10 cm in length and wind dispersed (Slik, 2009 onwards). It is known to be pollinated by the widespread giant honey bee Apis dorsata (Appanah, 1991). A. dorsata are colonial, with nests that may be densely congregated (often on adult Koompassia trees) and contain tens of thousands of individuals. Although it is possible for A. dorsata to nest in urban areas, when colonies in Singapore are discovered in urban areas, they are removed without exception. In Singapore there have been direct observations of A. cerana and various stingless bee species visiting K. malaccensis (JXQ Lee, personal communication), and it may also be pollinated by larger-bodied Xylocopa spp. (J Ascher, personal communication). Carpenter bees (Xylocopa spp.) tend to be solitary and most often nest in dead wood cavities, and in Singapore the two likely long-distance flying Xylocopids are Xylocopa latipes and X. flavonigrescens.
Study sites and sampling
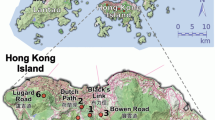
This study was conducted in three sites that contain (to our knowledge) all but a few remaining K. malaccensis adults in Singapore: Bukit Timah Nature Reserve (BT), MacRitchie Reservoir Park (MR, part of the Central Catchment Nature Reserve) and the Singapore Botanic Gardens (BG) (Figure 1). These three sites also retain the vast majority of extant primary rainforest (Yee et al., 2011). BT is a 163 ha forest fragment consisting of a single ~50 ha contiguous primary forest core surrounded by a secondary forest buffer. MR is a forest reserve consisting of a (relatively large) secondary forest fragment with small primary forest fragments embedded therein; in total, ~81 ha of primary forest were contained in five fragments of 6.9, 11, 13 and 40 ha, along with four very small fragments of 0.8–1.8 ha (Figure 1). Distances between the primary forest fragments in MR, and therefore the reproductive adults found in those fragments, ranged from <100 m to ~800 m. BG has one ~6 ha remnant of primary forest within which three adults were found; two other adults are isolated individuals outside the forest patch but within the Garden boundaries. The matrix between BG and the other patches consist largely of urban housing and built-up areas (Figure 1). Between MR and BT is a narrow corridor of young secondary forest, a six-lane highway, a pipeline and grassland.

Inset map: Singapore’s remaining primary forest areas and the sample sites in this study (black) and surrounding urban matrix (light gray). Main map: Distribution of the 179 sampled adult Koompassia malaccensis individuals (red dots) in the Botanic Gardens (N=5), Bukit Timah (N=52) and MacRitchie (N=122). Dark green is primary forest; light green is secondary forest; blue is water (from Noreen and Webb, 2013).
This study utilizes the conceptual and analytical term ‘patch’ to refer to clusters of K. malaccensis trees; mechanistically, we assumed that trees within a patch were more likely to outcross with others in that same patch, than with trees in another patch (see Figure 1). Although it would be possible to break the MR patch into smaller clusters, the analysis here focuses on broader patterns of gene flow. Between all patches there is, or has been, substantial habitat modification, thus isolating patches by both distance and habitat permeability (Kupfer et al., 2006).
We calculated patch area as the total area of a convex polygon circumscribing all Koompassia adults at a site (drawn in Google Earth and calculated using Earth Point, http://www.earthpoint.us/Shapes.aspx). Total patch sizes were 375 ha (MR), 84 ha (BT) and 1.4 ha (BG) (Table 1). Distances between the centroids of each patch were 4.8 km (BT–MR), 6.3 km (BT–BG) and 4.0 km (MR–BG). As our definition of patch specifically refers to the spatial extent of the tree clusters, patches could therefore include multiple habitat types, most commonly primary and secondary forest.
Detailed field sample collection methods, microsatellite genotyping and basic genetic data for K. malaccensis within Singapore have been summarized in Noreen and Webb (2013). Field surveys were conducted from November 2010 to May 2012. Across all sites, tissue samples from a total of 179 adults (⩾30 cm stem diameter) were collected as either freshly fallen leaves collected on the ground beneath an isolated adult, or as a 1 × 1 cm bark scraping (<4 mm deep) if another adult was within 50 m. BG contained five adults (Table 1). In BT, 52 of 54 known adult trees were located using a map constructed from a previous field study by SKY Lum (Table 1). We are confident that every adult K. malaccensis in the BG and >95% of the adults in BT were sampled; all adult trees are known and identified in the BG, and every tree with a diameter at breast height of >30 cm in BT had been previously tagged and identified by a project run by one of the co-authors of this study (SKY Lum). Adults in MR were located through several weeks of reconnaissance hikes, using the forest habitat map of Yee et al. (2011) as a guide to locate primary forest. In MR, 122 adults were located and sampled (Table 1). The position of each adult was recorded with a handheld GPS.
We collected a single leaflet from recruits up to ~30 m from the tree, keeping 3 m between sampled recruits whenever possible. For isolated adults with few seedlings, sample collection occurred up to 70 m. The number of seedlings within 70 m of adults ranged from many hundreds to zero. In most cases, there was an excess of seedlings to sample under the adult; for low-density cohorts all seedlings were sampled. We genotyped 101 recruits sampled from below 4 of the 5 adult trees at BG (no recruits were found under one of the adults isolated in the garden area); samples per tree ranged from 18 to 31 (mean=25.5 per adult). In BT we genotyped 812 recruits from under 47 (90%) of the 52 adults in BT (no recruits were found for 4 adults, and 1 adult had a single recruit that was unable to amplify); samples per tree ranged from 1 to 20 (mean=17.3 per adult). In MR, we genotyped 1100 recruits from under 70 (57%) of the 122 adults, ranging from 2 to 21 per tree (mean=15.7 per adult). Aside from variation in fecundity, differences in sampling intensity were due to the financial and logistical constraints of field work and genotyping.
Recruits were classified as >1 year old (55 cm to +2 m in height, n=193) or <1 year old (<50 cm, often with cotyledons still attached, n=1822). Detailed genetic diversity indices from Noreen and Webb (2013) showed no significant differences among age classes, and because of having only 193 recruits that were >1 year old among the three patches, all recruits were combined into a single category. Each sampled recruit was precisely mapped by measuring the distance (using a laser distance meter) and compass direction (Suunto KB-14, Suunto, Vantaa, Finland) to the nearest Koompassia adult.
Molecular methods
DNA extraction, PCR and genotyping followed methods in Noreen and Webb (2013). Eight highly robust microsatellite markers (Lee et al., 2006) were fluorescently labeled (VIC, FAM, NED or PET: Applied Biosystems, Carlsbad, CA, USA) and grouped into two multiplex PCR reactions of four markers each. One-third of adults and 0.7% of the recruit cohorts were re-extracted, PCRed and genotyped in their entirety, and 100% of adults and 24.9% of the recruit cohorts were re-PCRed and genotyped at one to eight loci to confirm rare alleles as well as to estimate genotyping errors. Genotyping errors were calculated per locus as the percent of nonmatching alleles in a subsequent PCR and ranged from 0.1 to 0.4% with a mean of 0.2% over the 8 loci. Missing data per locus comprised 0.1 to 4.3% with a mean of 1.0% over the 8 loci (see Noreen and Webb, 2013 for details). Given that all adults were re-extracted and genotyped in their entirety a minimum of twice, in addition to the low amount of missing data and low genotyping error rates, we are confident that there are few technical sources of error in the data set.
Data analysis: genetic diversity and null allele correction
GenePop v. 4.2 (Rousset, 2008) was used to calculate the observed and expected heterozygosities (Ho and He) and inbreeding coefficients (FIS); Fstat v. 2.9.3 was used to calculate allelic richness (R) (Goudet, 2001) (Supplementary Table S1). Microchecker v. 2.2.3 (van Oosterhout et al., 2004) was used to detect the presence of null alleles and possible technical scoring errors. Locus Km071 was detected to have significant levels of null alleles (~30%); Km141 and Km143 had low levels of null alleles (~5%). Locus Km071 was corrected for null alleles based on Chapuis and Estoup (2007). Effectively, Microchecker estimates the number of apparent homozygous individuals in a population that are actually heterozygotes containing a null allele. Using this information, we randomly chose the appropriate number of individuals homozygotic at Km071 for each patch, and replaced one allele with an allele size of 500. Although this method does not allow for particular individuals to be specifically pinpointed to have a null allele, it does allow for the correct proportion of homozygous individuals to be made into heterozygotes. Using the highest number of loci possible is critical for parentage analysis (Harrison et al., 2012); this rationale justifies this technique as a solution to retain this highly polymorphic locus (N=23 alleles). All data analysis beyond the genetic indices described above used data corrected for null alleles at Km071.
Data analysis: parentage assignment
Using Cervus v. 3.0.3 (Marshall et al., 1998), we conducted parentage analysis according to the following protocol: first, the allele frequency file was created from the existing adult data and simulations conducted; parent pair (sexes unknown), 100 000 offspring, 200 candidate parents, 90% candidate parents sampled, 0.99 typed, 0.01 mistyped, confidence LOD (logarithm (base 10) of odds) 95% relaxed and 99% strict, with possible self-fertilization and inbreeding. The allele frequency and simulations were then used as the basis of the analyses with all 179 adults retained as possible parents. The exclusion probability for excluding a putative parent pair (neither parent known) was 0.9999999938. The most likely parent pair was determined using most-likely joint LOD scores; recruits could have no parent assigned at P<0.05 (4.2% of recruits), or only one parent assigned at P<0.05 or P<0.01 (19.3% of recruits) or both parents assigned at P<0.05 or P<0.01 (75.6% of recruits) (see Supplementary Table S2 for a detailed breakdown). Parentage analysis results shown are from the 1524 recruits with both parents assigned: 1130 recruits at P<0.01 and 394 recruits at P<0.05.
We assumed that the assigned parent that was physically closest to the seedling was the maternal tree, with pollen dispersal distance calculated as the Euclidean distance between the two assigned parent trees. We defined an interpatch dispersal event as when one (putative pollen dispersal) or both (putative seed dispersal) parent(s) were from outside the patch in which the recruit was sampled. When the same adult was assigned as both maternal and paternal parent for a recruit, we classified the seedling as selfed.
Data analysis: immigration using the spatially explicit neighborhood model
Pollen and seed immigration into each patch was estimated using the spatially explicit seedling neighborhood model (Burczyk et al., 2006) implemented in the program NM+ (Chybicki and Burczyk, 2010). Using maximum likelihood, NM+ calculates parentage probabilities for mapped and genotyped progeny in relation to mapped and genotyped candidate parents. As opposed to parentage analysis, which is blind to geographical location of adults and cannot distinguish the maternal vs paternal contribution to the seedling, the neighborhood model determines the probable genealogy of seedlings taking into account: (1) the two-stage dispersal process (first pollen disperses from paternal to maternal tree, then seeds disperse away from maternal tree) and (2) the geographic proximity to candidate parents (the ‘neighborhood’). Pollen and seed immigration were estimated simultaneously, for each patch and for the data set as a whole (all Singapore). Settings used were: infinite neighborhood (to include all candidate parents), stop criterion 0.001, exponential-power dispersal kernel, genotyping error rates 0.1–0.4% per locus (Noreen and Webb, 2013), with the exceptions of Km071 (5% error rate used) and Km141 and Km143 (1% error rate used) to help account for null alleles, and default setting for the other parameters.
Data analysis: SGS
SGS autocorrelation plots were generated in GenAlex v. 6.5 (Peakall and Smouse, 2006) using Queller and Goodnight’s relatedness coefficient (r) (Queller and Goodnight, 1989). User-defined even-distance classes were assigned up to the longest distance class within each patch. Significance was calculated by: (1) shuffling the geographic locations of individuals (9999 permutations) to obtain the 95% confidence interval bars and (2) bootstrapping pairwise comparisons for each distance class to obtain the 95% confidence envelope around mean r values for distance classes (9999 permutations).
Results
Genetic diversity (allelic richness and expected heterozygosity) was high in recruits in all patches, and in the adults in BT and MR (sample size prevented calculation of these parameters for BG adults) (Table 2). Recruits had more alleles detected than the adults in each patch, but statistical analyses were not adequately robust to draw significant conclusions (Table 2). Outcross pollen limitation was likely in smaller patches with fewer adults, as shown by an increase in selfing rates as a proportion of the total seedlings, and also the percent of adult trees with at least one self-fertilized recruit (Table 3). Parentage analysis showed that ∼97% of the recruits were located within 100 m of the putative maternal tree, and this was expected given the sampling design and the fact that K. malaccensis seeds are wind dispersed. The parentage analysis detected 40 instances (2.6%) of putative seed dispersal events of 100–1000 m, and 5 instances (0.3%) of >1000 m. Of these 45 instances (involving 29 trees), 2 were from BG, 21 from BT and 22 from MR.
Pollen dispersal curves exhibited a higher frequency of short-distance pollen dispersal compared to adult pairwise distances (Figure 2), with median pollen dispersal distances of 163.8 m (BG), 186.9 m (BT) and 143.1 m (MR) (Table 3), which are shorter than median distances between adults within a patch (BG=298.7, BT=476.6 and MR=844.8; Table 1) and more aligned with near-neighbor distances. However, long-range pollen dispersal distance was evident (Figure 2), as we detected 794 instances (51.8%) of pollen dispersing 100–1000 m, and 112 instances (7.3%) of pollen dispersing >1000 m, and maximum pollen distances of 5.5–6.4 km (Table 3). BG had the highest proportion of recruits with outside-patch parents (both parental trees assigned; 13.6 vs 4.7 and 4.1%, independent samples Kruskal–Wallis test across sites, P<0.01, Table 3). Via parentage analysis, we detected 76 putative interpatch pollen dispersal events (in all directions between all patches, except from BG to BT; Figure 3) compared with just two putative interpatch seed dispersal events (both to BG).

The pairwise distance between all adult Koompassia malaccensis trees in each patch (black bars); the pairwise distance between the assigned parents (parentage analysis) of seedlings in that patch (gray bars). Distances are rounded to the nearest 100 m below 1050 m, and binned to the nearest 500 m beyond 1050 m.

Inter-patch gene flow of Koompassia malaccensis in the three main primary forest patches in Singapore based on parentage analysis. Each line represents the movement of pollen (76 events) or seeds (two events) between patches, with the source being (a) MacRitchie, (b) Bukit Timah and (c) the Botanic Gardens.
Parentage analysis assigned 93% of sampled adult K. malaccensis as a parent to at least one seedling. Of the adults, 87% were detected as pollen donors, of which approximately one-third (31%) fathered one to three sampled recruits. The top-producing five trees (3% of the adults) sired 27% of all sampled recruits for which both parents were assigned.
The neighborhood model for each site estimated substantially higher pollen immigration rates than did parentage analysis, but similar seed immigration rates (Table 4). Of the three patches, BG had the highest estimated pollen and seed immigration (pollen 0.459 (s.e. 0.053); seed: 0.058 (s.e. 0.025)); BT had the lowest (pollen 0.214 (s.e. 0.017); seed: 0.005 (s.e. 0.003)) (Table 4). For the whole data set (all of Singapore), the pollen immigration estimate was 0.349 (s.e. 0.012), whereas seed immigration was 0.023 (s.e. 0.004).
The SGS of all cohorts (adults, and the recruits from each of the three patches) showed significant structure (P<0.001; Figure 4 and Supplementary Figure S1c), with the individuals located in the first distance class (0–25 m) having mean relatedness (r) values of 0.139 (adults), 0.170 (BG), 0.226 (MR) and 0.245 (BT). The SGS of MR recruits, BT recruits and adults was not significantly different up to 500 m (P>0.05). However, the SGS of the BG recruit cohort was different from all other distributions, exhibiting strong positive relatedness within 100 m and strong negative relatedness at nearly all distances >100 m (Figure 4).

Overlaid SGS autocorrelation plots for up to 500 m, even-distance classes in 25 m intervals, for all Koompassia malaccensis adults (light blue, N=179), MacRitchie recruits (red, N=1100), Bukit Timah recruits (green, N=812) and Botanic Garden recruits (purple, N=101); r, relatedness value of Queller and Goodnight (1989). Bars are 95% confidence intervals (CIs). Individual patch autocorrelation plots up to the maximum distance class within each patch are shown in Supplementary Figures S1a–c.
Discussion
To our knowledge, this is the first study of gene flow of insect-pollinated trees in multiple patches in an urban landscape. Four key results parallel published results from insect-pollinated trees sampled in agricultural/mixed-forest landscapes. First, insects were able to transfer pollen across many km (>6 km), even across considerable stretches of ‘impervious’ substrates (~2.5 km); this agrees with the results of a review in Dick et al. (2008). Second, we detected relatively more frequent long-distance gene flow among isolated tree stands than in more continuous and larger forest patches (see Aldrich and Hamrick, 1998; Lander et al., 2010; Fuchs and Hamrick, 2011; Ismail et al., 2012; Rymer et al., 2013; Tambarussi et al., 2015; Guidugli et al., 2016). Third, specific individuals had a disproportionate contribution to gene flow among patches (see Aldrich and Hamrick, 1998; Fuchs and Hamrick, 2011; Lander et al., 2010; de Moraes and Sebbenn, 2011; Ismail et al., 2012); for example, a single tree in MR was a pollen donor for four different trees in BT as well as one in BG; and two BT trees received pollen from a total of 11 MR pollen donors. Finally, self-fertilization rates increased as patch size became smaller and adult abundance declined (see Ferreira et al., 2013). These four key findings demonstrate that insect-mediated gene flow patterns among patches (and by inference, pollinator behavior) can persist across a wide range of landscape types and configurations.
The significant SGS structure for all cohorts supports our findings of a high frequency of short-distance pollination and routine limited seed dispersal distances (that is, <100 m) that has resulted in significant relatedness among individuals at shorter distance classes. Because there was no difference in the SGS among the adults, the MR recruits and the BT recruits, we conclude that the combined pollen and seed dispersal dynamics within the two largest patches are relatively similar to each other; in addition, the result suggests that current gene flow dynamics of these two sites remain similar to historic levels, that is, when the adults were recruits.
In contrast, the BG recruits exhibited a significantly different SGS despite having median pollen and seed dispersal distances similar to MR and BT. Very high relatedness within 100 m and very negative relatedness beyond 100 m suggest a strong bimodal BG recruit cohort. For many of the BG recruits, both parents were within the patch (~48% neighborhood model estimate, ~67% parentage analysis theoretical estimate). As determined by parentage analysis, ~31% of the total number of BG recruits were selfed (N=25), and for the outcrossed seedlings whose parents were both from BG (N=46), they were the products of 1 of only 7 within-patch parent pairs (that is, we detected 7 within-BG parent pairs out of a possible maximum of 25). These reproductive dynamics within the patch contributed to high levels of relatedness for the entirely locally derived recruits. On the other hand, long-distance gene flow represented up to ~33% (parentage analysis) to ~52% (neighborhood model) of the BG recruit cohort. Of these recruits, parentage analysis (when both parents were assigned) captured 10 sampled outside-patch parental trees, all of which are geographically distant from each other and provided diverse immigrant genes. Hence, a bimodal cohort structure resulted in a SGS that was very different compared with the other patches, with marked fluctuations and some strongly positive relatedness values because of the within-patch reproductive dynamics, and some strongly negative values owing to the high proportion of immigrant genes unrelated to each other or to the local adults.
In light of the generally low estimates of long-distance seed dispersal, the elevated seed immigration rate for BG was unexpected, given that seeds would have to be dispersed across a minimum of 2.5 km of urban habitat. There was also no evident trend to explain why particular maternal trees would be geographically well situated for long-distance dispersing seeds. It is possible that some mother trees remained undiscovered by our sampling: some BG recruits had two non-matching alleles for particular loci compared with the five BG adults, and the non-matching alleles had different identities among these recruits. This result is unlikely because of mistyping or mutation, and therefore suggests that there were multiple unknown mother trees.
The variance in fecundity we detected suggests both positive and negative long-term implications for K. malaccensis in Singapore. On one hand, almost all adults had contributed genes to at least one recruit, indicating maintenance of allelic richness and rare alleles contained in the extant population of genetically diverse Koompassia adults. This contrasts with other studies that have detected significantly lower allelic richness in recruits of small remnant populations (see, for example, Kettle et al., 2007; Finger et al., 2012; Guidugli et al., 2016) that may be attributed to the loss of rare alleles owing to non-reproductive adults and/or unequal fecundity (see Finger et al., 2012). On the other hand, parentage analysis detected that five trees sired approximately a quarter of the genotyped seedlings: this inequality, if continued over subsequent years, could contribute to genetic drift and produce negative fitness effects in future generations. However, natural variation in adult fecundity across reproductive cycles could average out the relative contributions of individuals to the overall recruit landscape over the long term; extending the present study across several reproductive cycles would reveal whether certain individuals are consistently top sires or whether their relative contributions change over time.
We documented increased selfing rates in smaller patches. Increasing self-fertilization and unequal fecundity can act synergistically, leading to inbreeding depression that could have profound fitness implications in the long term (see, for example, Breed et al., 2012; Rymer et al., 2013; Porcher and Lande, 2016). Correlated paternity rates could increase in the future: with a few individuals siring a relatively large proportion of offspring, more recruits are likely to be half-siblings. SGS shows that related individuals are geographically close to each other—even for adults—and that near-neighbor matings are common. Hence, the selfing rate and unequal fecundity, in conjunction with sustained biparental inbreeding, could have a detrimental fitness effect in subsequent generations (see, for example, Breed et al., 2012; Rymer et al., 2013; reviewed in Ghazoul, 2005). However, the century-long lifespan of K. malaccensis individuals and the management of Singapore’s urban forest patches (see, for example, lightning rods installed on many large trees, extending their lifespan) has likely prevented the detrimental accumulation of inbreeding depression over generations: a large percent of the seedling recruit cohort we sampled are effectively still the ‘first generation post-fragmentation’ despite the bulk of fragmentation occurring >120 years previously.
This study has several limitations that temper our conclusions. The study was conducted on only three patches, with different areas and adult abundances as well as different types of surrounding matrix. Thus, it was not possible to fully disentangle all independent effects. We also sampled recruits from under only 57% of MR adults because of logistical and financial constraints; however, as 93% of MR adults were assigned as parents, we believe our sampling strategy was successful in capturing gene flow. In addition, in 2015–2016, one of the authors (MA Niissalo) spent ~50 h surveying the area containing the tiny primary forest patches ~4 km north of the northernmost MR tree (see Figure 1); he positively identified three adult K. malaccensis (diameter at breast height ~40–50 cm each) with seedlings, in or very close to the tiny remaining primary forest patches. Hence, given the number of reproductive adults detected for this survey effort, the small number of tiny primary forest patches remaining and the immigration estimates of ca. 2–3% (seed) and ca. 20–35% (pollen) for the whole data set, we conclude that there are further unsampled adults in the northern areas of the Central Catchment but that these adults are likely very few in number.
The core question of this study has a clear overall conclusion: substantial insect-mediated gene flow between trees in remnant forest patches was maintained in this highly urbanized landscape, despite the ‘impervious’ matrix. Inconsistency in estimates of pollen flow based on parentage analysis versus the neighborhood model prevents a precise estimation of the magnitude of pollen dispersal; an in-depth analysis of the two models could be conducted but is beyond the scope of this paper. Moreover, subtle differences in dispersal between MR and BT are observed when BT is the pollen source compared with when MR is the source (see Figure 3), suggesting asymmetrical source–sink dynamics. It would have been helpful to identify all species of pollinator for K. malaccensis and differences (if any) in visitation rate among patches, as without this information many important questions about the pollinator behavior remain unanswered. For example, it would be interesting to determine whether short-range pollen dispersal is performed by same species as is the long-distance dispersal, and whether urbanization affects the abundance and/or behavior of some pollinator species more profoundly than others. The large carpenter bee X. flavonigrescens is a very strong flier and is genetically panmictic throughout Singapore (including islands up to 4.5 km offshore; Yi, 2015), but the extent to which this species contributes to the long-distance pollen dispersal among patches is unknown. Hence, future studies on insect-mediated dispersal should attempt to quantify, to at least some extent, the identity and frequency of pollinator visitation among patches.
Finally, the substantial gene flow we detected among primary forest patches in an urban setting for K. malaccensis are likely to be a ‘best-case’ scenario: it is a long-lived, previously widespread canopy tree species pollinated by strong fliers. Studies are needed for rare or understory plant species, and plants pollinated by less mobile and/or less abundant animals, both of which would be predicted to have lower levels of connectivity than K. malaccensis among forest patches in the urban landscape.
Data archiving
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.663fb.
References
Aguilar R, Quesada M, Ashworth L, Herreriasdiego Y, Lobo J . (2008). Genetic consequences of habitat fragmentation in plant populations: susceptible signals in plant traits and methodological approaches. Mol Ecol 17: 5177–5188.
Aldrich PR, Hamrick JL . (1998). Reproductive dominance of pasture trees in a fragmented tropical forest mosaic. Science 281: 103–105.
Alvey AA . (2006). Promoting and preserving biodiversity in the urban forest. Urban For Urban Green 5: 195–201.
Appanah S . (1991) Plant–pollinator interactions in Malaysian rain forests. In: Bawa KS, Hadley M (eds), Reproductive Ecology of Tropical Forest Plants. International Union of Biological Sciences Special Issue 21 (Biology International): Paris, France. pp 85–102.
Breed MF, Marklund MHK, Ottewell KM, Gardner MG, Berton J, Harris BC et al. (2012). Pollen diversity matters: revealing the neglected effect of pollen diversity on fitness in fragmented landscapes. Mol Ecol 21: 5955–5968.
Burczyk J, Adams WT, Birkes DS, Chybicki IJ . (2006). Using genetic markers to directly estimate gene flow and reproductive success parameters in plants on the basis of naturally regenerated seedlings. Genetics 173: 363–372.
Chapuis MP, Estoup A . (2007). Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24: 621–631.
Chybicki IJ, Burczyk J . (2010). NM+: software implementing parentage-based models for estimating gene dispersal and mating patterns in plants. Mol Ecol Res 10: 1071–1075.
Corlett RT . (1992). The ecological transformation of Singapore, 1819-1990. J Biogeogr 19: 411–420.
Davis ES, Murray TE, Fitzpatrick U, Brown MJF, Paxton RJ . (2010). Landscape effects on extremely fragmented populations of a rare solitary bee, Colletes floralis. Mol Ecol 19: 4922–4935.
de Moraes MLT, Sebbenn AM . (2011). Pollen dispersal between isolated trees in the Brazilian savannah: a case study of the neotropical tree Hymenaea stigonocarpa. Biotrop 43: 192–199.
DeFries RS, Rudel T, Uriarte M, Hansen M . (2010). Deforestation driven by urban population growth and agricultural trade in the twenty-first century. Nat Geosci 3: 178–181.
Delaney KS, Riley SPD, Fisher RN . (2010). A rapid, strong, and convergent genetic response to urban habitat fragmentation in four divergent and widespread vertebrates. PLoS One 5: e12767.
Dick CW, Hardy OJ, Jones FA, Petit RJ . (2008). Spatial scales of pollen and seed-mediated gene flow in tropical rain forest trees. Trop Plant Biol 1: 20–33.
Eckert CG, Kalisz S, Geber MA, Sargent R, Elle E, Cheptou PO . (2010). Plant mating systems in a changing world. Trends Ecol Evol 25: 35–43.
Ferreira PA, Boscolo D, Viana BF . (2013). What do we know about the effects of landscape changes on plant–pollinator interaction networks? Ecol Indic 31: 35–40.
Finger A, Kettle CJ, Kaiser-Bunbury CN, Valentin T, Mougal J, Ghazoul J . (2012). Forest fragmentation genetics in a formerly widespread island endemic tree: Vateriopsis seychellarum (Dipterocarpaceae). Mol Ecol 21: 2369–2382.
Fuchs EJ, Hamrick JL . (2011). Mating system and pollen flow between remnant populations of the endangered tropical tree, Guaiacum sanctum (Zygophyllaceae). Conserv Genet 12: 175–185.
Gao Q, Yu M . (2014). Discerning fragmentation dynamics of tropical forest and wetland during reforestation, urban sprawl, and policy shifts. PLoS One 9: e113140.
Ghazoul J . (2005). Pollen and seed dispersal among dispersed plants. Biol Rev 80: 413–443.
Goudet J . (2001). FSTAT, Version 2.9.3. A Program to Estimate and Test Gene Diversities and Fixation Indices. Lausanne University: Lausanne, Switzerland.
Guidugli MC, Nazareno AG, Feres JM, Contel EPB, Mestriner MA, Alzate-Marin AL . (2016). Small but not isolated: a population genetic survey of the tropical tree Cariniana estrellensis (Lecythidaceae) in a highly fragmented habitat. Heredity 116: 339–347.
Harrison HB, Saenz-Agudelo P, Planes S, Jones GP, Berumens ML . (2012). Relative accuracy of three common methods of parentage analysis in natural populations. Mol Ecol 22: 1158–1170.
Hitchings SP, Beebee TJC . (1997). Genetic substructuring as a result of barriers to gene flow in urban Rana temporaria (common frog) populations: implications for biodiversity conservation. Heredity 79: 117–127.
Ismail SA, Ghazoul J, Ravikanth G, Shaanker UR, Kushalappa CG, Kettle CJ . (2012). Does long-distance pollen dispersal preclude inbreeding in tropical trees? Fragmentation genetics of Dysoxylum malabaricum in an agro-forest landscape. Mol Ecol 21: 5484–5494.
Jha S, Kremen C . (2013). Urban land use limits regional bumble bee gene flow. Mol Ecol 22: 2483–2495.
Keller I, Largiadèr CR . (2003). Recent habitat fragmentation caused by major roads leads to reduction of gene flow and loss of genetic variability in ground beetles. Proc R Soc Lond Ser B 270: 417–423.
Kettle CJ, Hollingsworth PM, Jaffré T, Moran B, Ennos RA . (2007). Identifying the early genetic consequences of habitat degradation in a highly threatened tropical conifer, Araucaria nemorosa Laubenfels. Mol Ecol 16: 3581–3591.
Kupfer JA, Malanson GP, Franklin SB . (2006). Not seeing the ocean for the islands: the mediating influence of matrix-based processes on forest fragmentation effects. Glob Ecol Biogeogr 15: 8–20.
Lander TA, Boshier DH, Harris SA . (2010). Fragmented but not isolated: contribution of single trees, small patches and long-distance pollen flow to genetic connectivity for Gomortega keule, an endangered Chilean tree. Biol Conserv 143: 2583–2590.
Lee CT, Lee SL, Faridah QZ, Siraj SS, Ng KKS, Norlia B et al. (2006). Isolation and characterization of microsatellite markers in Koompassia malaccensis (Leguminosae), an important tropical timber species. Mol Ecol Notes 6: 1198–1201.
Lee CT, Lee SL, Ng KKS, Faridh QZ, Siraj SS, Norwati M . (2011). Estimation of outcrossing rates in Koompassia malaccensis from an open-pollinated population in peninsular Malaysia using microsatellite markers. J Trop For Sci 23: 410–416.
Marshall TC, Slate J, Kruuk LEB, Pemberton JM . (1998). Statistical confidence for likelihood-based paternity inference in natural populations. Mol Ecol 7: 639–655.
Munshi-South J, Kharchenko K . (2010). Rapid, pervasive genetic differentiation of urban white-footed mouse (Peromyscus leucopus populations in New York City. Mol Ecol 19: 4242–4254.
Nagamitsu T, Kikuchi S, Hotta M, Kenta T, Huira T . (2014). Effects of population size, forest fragmentation, and urbanization on seed production and gene flow in an endangered Maple (Acer miyabei. Am Midl Nat 172: 303–316.
Noël S, Oullet M, Galois P, Lapointe F-J . (2007). Impact of urban fragmentation on the genetic structure of the eastern red-backed salamander. Conserv Genet 8: 599–606.
Noreen AME, Webb EL . (2013). High genetic diversity in a potentially vulnerable tropical tree species despite extreme habitat loss. Plos One 8: e82632.
Ollerton J, Winfree R, Tarrand S . (2011). How many flowering plants are pollinated by animals? Oikos 120: 321–326.
Peakall R, Smouse PE . (2006). GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6: 288–295.
Porcher E, Lande R . (2016). Inbreeding depression under mixed outcrossing, self-fertilization and sib-mating. BMC Evol Biol 16: 105.
Queller DC, Goodnight KF . (1989). Estimating relatedness using genetic markers. Evolution 43: 258–275.
Rousset F . (2008). Genepop'007: a complete reimplementation of the Genepop software for Windows and Linux. Mol Ecol Res 8: 103–106.
Rymer PD, Sandiford M, Harris SA, Billingham MR, Boshier DH . (2013). Remnant Pachira quinata pasture trees have greater opportunities to self and suffer reduced reproductive success due to inbreeding depression. Heredity 115: 115–124.
Seamon J, Mann S, Steele OC, Utzurrum RB . (2006). Conservation value of remnant forest patches: tree composition, spatial patterns, and recruitment in the Ottoville Lowland Forest, American Samoa. Pac Sci 60: 319–332.
Sebbenn AM, Carvalho ACM, Freitas MLM, Moraes SMB, Gaino APSC, da Silva JM et al. (2011). Low levels of realized seed and pollen gene flow and strong spatial genetic structure in a small, isolated and fragmented population of the tropical tree Copaifera langsdorffii Desf. Heredity 106: 134–145.
Slik JWF . (2009 onwards). Plants of Southeast Asia http://www.asianplant.net Accessed 4 July 2016.
Tambarussi EV, Boshier D, Vencovsky R, Freitas MLM, Sebbenn AM . (2015). Paternity analysis reveals significant isolation and near neighbor pollen dispersal in small Cariniana legalis Mart. Kuntze populations in the Brazilian Atlantic Forest. Ecol Evol 5: 5588–5600.
Van Oosterhout C, Hutchison WF, Shipley P, Wills DPM . (2004). Micro-Checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol 4: 535–538.
Wang H, Sork V, Wu J, Ge J . (2010). Effect of patch size and isolation on mating patterns and seed production in an urban population of Chinese pine (Pinus tabulaeformis Carr.). For Ecol Manag 260: 965–974.
Yee ATK, Corlett RT, Liew SC, Tan HTW . (2011). The vegetation of Singapore—an updated map. Gard Bull Singapore 63: 205–212.
Yi NK . (2015) Population genomics of cryptic carpenter bee species (Hymenoptera: Apidae: Xylocopa) in Singapore. Final Year Project, National University of Singapore, Singapore.
Acknowledgements
We thank Lahiru Wijedasa, Wei Kit Lee, Alison Wee, Rachel Webb, John Ascher and Professor Rudolf Meier in addition to the many volunteers who aided in field work. Collections were undertaken under Singapore National Parks Research Permit NP/RP10-078. We thank Alex TK Yee for providing assistance with Figure 1. The study was supported by the Singapore Ministry of Education (Grant R-154-000-400-133) and the Lee Hiok Kwee Endowment to the Department of Biological Sciences, NUS (Grant R-154-000-480-720).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Rights and permissions
About this article

Cite this article
Noreen, A., Niissalo, M., Lum, S. et al. Persistence of long-distance, insect-mediated pollen movement for a tropical canopy tree species in remnant forest patches in an urban landscape. Heredity 117, 472–480 (2016). https://doi.org/10.1038/hdy.2016.64
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2016.64
This article is cited by
-
Pollen dispersal and mating patterns determine resilience for a large-yet-fragmented population of Cariniana estrellensis
Conservation Genetics (2024)
-
Population genetics of the European rabbit along a rural-to-urban gradient
Scientific Reports (2020)
-
The effects of pollen dispersal and mating pattern on inbreeding depression and hybrid vigor in Balfourodendron riedelianum (Engl.) Engl. (Rutaceae)
Conservation Genetics (2020)
-
Short distance pollen dispersal and low genetic diversity in a subcanopy tropical rainforest tree, Fontainea picrosperma (Euphorbiaceae)
Heredity (2019)
-
A fig tree in a concrete jungle: fine-scale population genetic structure of the cluster fig Ficus racemosa in an urban environment
Urban Ecosystems (2018)
