
Abstract
The combination of oceanographic barriers and habitat heterogeneity are known to reduce connectivity and leave specific genetic signatures in the demographic history of marine species. However, barriers to gene flow in the marine environment are almost never impermeable which inevitably allows secondary contact to occur. In this study, eight sampling sites (five along the South African coastline, one each in Angola, Senegal and Portugal) were chosen to examine the population genetic structure and phylogeographic history of the cosmopolitan bluefish (Pomatomus saltatrix), distributed across a large South-east Atlantic upwelling zone. Molecular analyses were applied to mtDNA cytochrome b, intron AM2B1 and 15 microsatellite loci. We detected uncharacteristically high genetic differentiation (FST 0.15–0.20; P<0.001) between the fish sampled from South Africa and the other sites, strongly influenced by five outlier microsatellite loci located in conserved intergenic regions. In addition, differentiation among the remaining East Atlantic sites was detected, although mtDNA indicated past isolation with subsequent secondary contact between these East Atlantic populations. We further identified secondary contact, with unidirectional gene flow from South Africa to Angola. The directional contact is likely explained by a combination of the northward flowing offshore current and endogenous incompatibilities restricting integration of certain regions of the genome and limiting gene flow to the south. The results confirm that the dynamic system associated with the Benguela current upwelling zone influences species distributions and population processes in the South-east Atlantic.
Similar content being viewed by others


Introduction
Identifying the processes driving population structuring and divergence within a species is essential for understanding its evolutionary dynamics. In the marine environment, high dispersal and limited genetic differentiation are often observed over large spatial scales (Palumbi, 1994; O’Reilly et al., 2004). However, a growing number of cases illustrate that divergence also occurs across limited geographic distances (Hauser and Carvalho, 2008; Hellberg, 2009). In addition, within the marine environment, barriers to gene flow that may lead to initial divergence are often not persistent, allowing sporadic gene flow between two populations to occur through time (Gaither et al., 2011). Due to the nature of most oceanographic barriers and the life history traits of many marine organisms (for example, benthopelagic life cycle) this sporadic gene flow leads to introgression of the neutral portions of the genome (Bierne et al., 2011, 2013).
A number of marine studies have found an association between high genetic differentiation (FST outliers) and differing ecological environments, which could arise through local adaptation to specific environments (exogenous barriers; Hemmer-Hansen et al., 2007; Larmuseau et al., 2009), or allopatric divergence with recent secondary contact (due to endogenous barriers; Bierne et al., 2011; Gagnaire et al., 2011), among others. Genetic divergence in the marine environment is often driven through transient physical barriers that arise due to sea-level changes and oceanographic barriers such as eddies, currents and upwelling zones, which are dependent on the climatic setting at a specific time (Cowen et al., 2000). These transient barriers allow gene flow to occur when conditions are favorable leading to high rates of introgression across these barriers (Bierne et al., 2011; Gagnaire et al., 2015). However, when strong genetic differentiation persists across such semi-permeable barriers, it is probable that processes other than genetic drift alone are limiting gene flow (Gagnaire et al., 2011; Quéré et al., 2012).
A number of recent marine studies have highlighted these processes, especially in hybrid zones or through evidence of past secondary contact (Gagnaire et al., 2009; Bierne et al., 2011; Quéré et al., 2012; Roux et al., 2013). These studies have identified heterogeneity of differentiation among loci, with few loci showing much larger divergence whereas others showed weak divergence or genetic homogeneity. These patterns are thought to form in tension zones which are the areas where populations of differing genetic backgrounds overlap and interbreed. These zones often coincide with natural barriers (Barton, 1979; Hewitt, 1988), or regions characterized by ecological gradients when exogenous and endogenous barriers become coupled (Bierne et al., 2011).
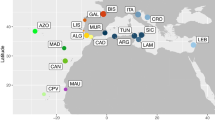
The heterogeneous features of the coastal waters of the East Atlantic (EA, Figure 1a) have the potential to affect genetic diversity and connectivity of marine organisms. Regional differences in the continental shelf depth and slope, temperature, salinity, apparent oxygen use, nutrient concentrations (for example, silicate, nitrate and phosphate; Silva et al., 2014b) and intense upwelling (Hutchings et al., 2009) have been observed. Two intense upwelling zones have been described; the first is an upwelling off the coast of Morocco/Mauritania, and the second is the cold Benguela upwelling on the border of South Africa and Namibia. There are two cold currents which flank the tropics in this region (the northward flowing Benguela current along southern Africa and the southward flowing Canary current in the North-east Atlantic; Figure 1). Along the South African coast specifically, the warm Agulhas current meets the Benguela current at the Agulhas Bank and forms the Indian/Atlantic Ocean transition zone which has been identified as a phylogeographic break in some coastal species (Teske et al., 2011, 2014). Also, the front between the Agulhas and Benguela currents has been shown to shift seasonally, leading to different positions of the hybrid zone in bigeye tuna (Durand et al., 2005a).

(a) Map of oceanographic features and sampling sites of Pomatomus saltatrix in the East Atlantic. Circles indicate sampling sites (Mediterranean—red; Portugal—orange, Senegal—light orange, Angola—yellow, South Africa—green). The arrows indicate the major currents and relative temperature: CC, Canary Current, NEC, North Equatorial Current, ECC, Equatorial Counter Current, GC, Guinea Current, AnC, Angolan Current, SEC, South Equatorial Current, BC, Benguela Current, AgC, Agulhas Current, the light blue shading indicates the upwelling zone at Lüderitz, Namibia. Haplotype networks based on (b) 1039 bp of cytochrome b and (c) 484 bp of AM2B1 with a 95% connection limit, the numbers in the circles represent the number of sequences to provide scale. (d, e) Two clusters were identified as the most likely based on all microsatellites, namely South Africa (SA) and the North East and Central Atlantic (Portugal (POR) and Senegal (SEN) and Angola (ANG, indicated in Orange)) using STRUCTURE 2.3.2. South African sampling sites are indicated in green and consist of Berg River (BR), False Bay (FB), East London (EL), Port Edward (PE) and St Lucia (SL).
Genetic studies on species from the East Atlantic region (not including South Africa) have identified several genetic breaks using mitochondrial DNA (mtDNA; Chikhi et al., 1998; Durand et al., 2005b). The sub-structuring described in the region was further characterized in Ethmalosa fimbriata, with four distinct nuclear genetic clusters identified (Durand et al., 2013). Studies focused on species distributed across the cold Benguela upwelling have reported divergences between South African populations and those located further north (Grant and Bowen, 1998; Teske et al., 2011; Henriques et al., 2012, 2014, 2015). Others found evidence of secondary contact of divergent South African haplogroups in the North-east Atlantic (Sala-Bozano et al., 2009).
The bluefish Pomatomus saltatrix is a species of particular interest in this region. It is a cosmopolitan coastal species occurring in most warm-temperate regions globally, and has the potential to disperse over large distances during both the pelagic larval stage and the vagile adult stage (Juanes et al., 1996). All life stages exhibit temperature-dependent behavior, with the adults of most populations migrating to warmer waters during seasonally cooler periods and preferentially spawning at temperatures ranging between 20 and 26 °C (Juanes et al., 1996; Sebastés et al., 2012). A previous broad-scale study based on mitochondrial restriction fragment length polymorphisms reported a close genetic relationship between populations of South Africa, Portugal and North America, in relation to populations occurring in Australia and Brazil (Goodbred and Graves, 1996). Additional studies in the Mediterranean and western North Atlantic have indicated sub-structuring and potential male-biased dispersal (Pardiñas et al., 2010; Miralles et al., 2014a, 2014b).
This species is distributed across several environmental features of the East Atlantic including both the major upwelling zones, as well as the Indian/Atlantic transition. Bluefish has a continuous distribution around the South African coastline and is found in both the cold-temperate and warm-temperate biogeographic regions. A large portion of this population has been observed to undergo an annual spawning migration from these habitats to the KwaZulu–Natal north coast (for example, St Lucia located in a warm-temperate biogeographic region; Figure 1) during the austral spring (Van der Elst, 1976).
In this study, we investigated the role of oceanographic barriers in shaping genetic differentiation in P. saltatrix across the EA, by including sampling from Portugal, Senegal, Angola and South Africa. We also evaluated the population genetic structure of the South African population across a known southern African phylogeographic break. To do so, we analyzed gene sequences (mtDNA cyt b and nuclear intron AM2B1), as well as a set of 15 microsatellite loci. We assessed the genetic patterns observed and quantified demographic parameters for this species along the West and southern African coastline.
Materials and methods
Sampling and laboratory procedures
Collections of P. saltatrix tissue were obtained from the EA and around the tip of South Africa into the Indian Ocean (Figure 1a). Sampling sites in South Africa included Berg River (n=29) and False Bay (n=38) in the EA, as well as East London (n=28), Port Edward (n=30) and St Lucia (SL; n=30) in the South Western Indian Ocean. Samples were also collected from three sites north of the Lüderitz upwelling zone: Angola (n=36), Senegal (n=11) and Portugal (n=16). Fin clips and muscle tissue were taken and stored in 95% ethanol or tissue storage buffer and then kept at −20 and 4 °C, respectively. DNA was extracted using the Qiagen DNeasy extraction kit according to the manufacturer’s specifications (Qiagen, CA, USA).
A 1078-bp fragment of the cyt b gene region (Supplementary Data S1, designed following the technique of Hoareau and Boissin, 2010) and a 488-bp region of alpha amylase intron 1 (AM2B1; Hassan et al., 2002) were sequenced in samples from all locations. The same polymerase chain reaction (PCR) and sequencing protocol was followed for both loci: PCR volumes of 25 μl included 100 ng of genomic DNA, 0.3 mM dNTPs (Promega, Madison, WI, USA), 0.5 U Supertherm Taq polymerase (Separation Scientific, Cape Town, RSA), 1 × PCR buffer (100 mM Tris-HCl, 500 mM KCl, pH 8.4), 1.5 mM MgCl2 and 2pmol of forward and reverse primers (Whitehead Scientific). The amplification protocol included a denaturation step of 94 °C for five minutes, and 35 cycles of 94 °C for 30 s, 45 s of annealing at 55 °C, extension of 72 °C for 45 s, and a final extension step of 72 °C for 7 minutes. PCR products were purified using Sephadex beads (Sigma-Aldrich), and cycle sequenced in both directions. Sequencing was done on an ABI3500xl automated sequencer (Applied Biosystems, Foster City, CA, USA). Six full length cyt b sequences already available for P. saltatrix from the North-east Atlantic/Mediterranean were downloaded from GenBank (accession numbers, sampling sites in Supplementary Table S1). Sequences were edited and assembled in CLC Bio Workbench 4.1.1 (CLC Bio A/S, Aarhus, Denmark). The various nuclear alleles for AM2B1 were reconstructed in DNASP 5.10 (Librado and Rozas, 2009) using the Bayesian PHASE algorithm (Stephens et al., 2001; Stephens and Donnelly, 2003). Two additional mtDNA regions, NADH Dehydrogenase 2 (ND2, primers, CJ Oosthuizen, unpublished data) and control region (CR; Chen et al., 2012) were sequenced in a small subset of samples following the above-mentioned protocol to clarify whether shared haplotypes identified in cyt b were more likely due to mtDNA introgression or ancestral polymorphism (Supplementary Figure S1).
The collected tissue was screened for variation at 15 polymorphic microsatellite loci (Supplementary Table S2). These included six microsatellites developed by Dos Santos et al. (2008), three newly developed loci from previous Fast Isolation by AFLP of Sequences COntaining repeats (FIASCO) enrichment (Zane et al., 2002) and cloning by Dos Santos et al. (2008), and six dinucleotide loci designed in conserved intergenic regions (IGRs) of four fish genomes (Gotoh et al., 2013). The loci were screened in three multiplex reactions and genotyped using the Qiagen Quantitect multiplexing kit (Qiagen) according to the manufacturer’s specifications (multiplexes, Supplementary Table S2) on an ABI3130 automated sequencer (Applied Biosystems).
Data analysis
MEGA 5 (Tamura et al., 2011) was used to align assembled sequences (CLUSTALW; Thompson et al., 1994), and to identify the best-fit mutation model for the cyt b data. The number of haplotypes (H), haplotype diversity (Hd), nucleotide diversity (π), estimates of θ based on the number of segregating sites (θS), mean pairwise differences (θπ) and neutrality tests (Fu’s Fs and Tajima’s D) were calculated for cyt b using ARLEQUIN 3.11 (Excoffier et al., 2005). Genetic differentiation (ΦST) among populations for cyt b and AM2B1 sequences was also calculated in ARLEQUIN. Haplotype networks were constructed based on statistical parsimony in TCS 1.2.1 (Clement et al., 2000) using a 95% connection limit.
Tests for null alleles were done with FREENA (Chapuis and Estoup, 2007). Summary statistics for the microsatellite loci were calculated with GENETIX 4.05 (Belkhir et al., 1996–2004), and included: the number of alleles (NA), observed (HO) and expected (HE) heterozygosity, and inbreeding coefficient (FIS; Weir and Cockerham, 1984), for each locus in each sampling site. Tests for deviations from Hardy–Weinberg equilibrium (HWE) among populations and linkage disequilibrium between loci were performed with GENEPOP (Rousset, 2008). When multiple statistical tests were performed, sequential Bonferroni correction was implemented (Rice, 1989). Tests for the presence of outlier loci among microsatellites were conducted with LOSITAN (Antao et al., 2008). Pairwise tests between sites were implemented for the step-wise mutation model (SMM; Kimura and Ohta, 1978), with 50 000 iterations and a 95% confidence interval.
To assess the spatial genetic structuring of P. saltatrix along the west coast of Africa, genetic differentiation (FST; Weir and Cockerham, 1984) was calculated with FREENA that provides FST values that are corrected for any potential null alleles using the excluding null alleles method (ENA; Chapuis and Estoup, 2007). These corrected estimates are expected to avoid any potential bias, as null alleles are known to be more frequent in high gene flow species such as most marine organisms (O’Reilly et al., 2004). Genetic differentiation was also calculated between specific sampling sites off the South African coast.
A Bayesian clustering method was implemented to determine the most likely number of independent populations (STRUCTURE 2.3.2; Pritchard et al., 2000). Several values of the number of populations (K=1–6) were tested, with 10 iterations of each K with a burn-in length of 300 000 followed by 300 000 Markov–Chain Monte–Carlo (MCMC) steps. The sampling site was specified as prior, with uncorrelated allele frequencies (Hubisz et al., 2009). The ad hoc statistic ΔK (Evanno et al., 2005) was used to confirm the most likely number of populations with STRUCTURE HARVESTER (Earl and VonHoldt, 2012).
To assess demographic parameters of P. saltatrix, the isolation-with-migration model (IMa) was implemented in IMa 1.0 (Hey and Nielsen, 2007). The full IMa model (including all parameters) was applied using all loci combined, with population 1 defined as the East Atlantic (including Portugal, Senegal and Angola) and population 2 as South Africa (input file, Supplementary Data S2). Additional IMa runs were done for each data set separately to characterize individual marker-type patterns across the Benguela upwelling zone (Angola vs South Africa). The IMa program uses a metropolis coupling MCMC method to estimate the posterior probability of various demographic parameters. The parameters estimated were the current effective population size of each population (θ1, θ2), ancestral population size (θA), the time of divergence (tsplit) and the direction and extent of gene flow (m1, m2) between descendent populations. To account for possible time-dependency of the rate of molecular evolution (Ho et al., 2005), a mutation rate of 9.4% per million years with a s.d. of 2.45% was used for cyt b based on a calibration derived from the calibration of demographic transition method using demographic processes of expansion of stickleback in the North Atlantic (Hoareau, 2016). A mutation rate of 1x10−5 was chosen for each microsatellite locus, which is within the known range of mutation rates for microsatellites (Selkoe and Toonen, 2006). Due to the lack of a known mutation rate for intron AM2B1, only the raw estimates are reported for this locus when run individually as they are not influenced by the mutation rate. The generation time used for P. saltatrix was 4.8 years (www.iucn.org). Parameters were set through several runs and included a burn-in of 2 × 106 and chain mixing with parameters –f g –n 40 –g1 0.6 –g2 0.85 for the combined data set and a burn-in of 1 × 108 and chain mixing (–ft –n5 –g1 0.05 –g2 2) for individual marker data sets. The accuracy of the various runs was established by having an effective sample size larger than 50 and no significant autocorrelations. Once the criteria were met, three independent iterations were done with random seeds for each data set. The input file for the combined data set is available in Supplementary Data S2.
Results
Sequence analysis of 86 individuals resulted in 37 cyt b haplotypes (GenBank accession numbers: KX196669–KX196754). Samples from South Africa had the lowest haplotype and nucleotide diversity compared with the other sampling sites and also had negative and highly significant values of Fu’s FS and Tajima’s D (Table 1). South Africa formed a haplogroup distinct from the sampling sites north of the Benguela upwelling zone, which comprised three haplogroups with shared haplotypes between them. The Angolan samples also shared three haplotypes with South Africa (15% of Angolan samples had South African haplotypes). Aside from these haplotypes, South Africa did not share haplotypes with other sampling sites (Figure 1b). Sequencing of CR and ND2 regions for the Angolan individuals sharing South African cyt b haplotypes allowed for the exclusion of ancestral polymorphism (Supplementary Figure S1). The nuclear intron (AM2B1) resulted in the analysis of 44 individuals (15 RSA, 29 NEA) and 14 unique haplotypes (GenBank accession numbers: KX196605-KX196648). No shared haplotypes between South Africa and the rest of the EA were observed at this locus. Among these other sites, Angola showed a number of private haplotypes (Figure 1c and Table 2).
The microsatellite loci showed no evidence of linkage disequilibrium. Two loci showed null alleles at several locations (although not all) and were removed from further analyses (Elf44, 7% and Elf37, 15% average across sampling locations). Standard FST estimates were highly correlated to FST corrected for the presence of null alleles (R=0.99, P<0.001), which indicates that null alleles did not significantly affect the conclusions of the study. However, we still report the corrected FST values in Table 2. Summary statistics for each marker and each sampling site are indicated in Supplementary Table S3. There were few deviations of genotypic distributions from the expectations of HWE. Using LOSITAN for pairwise comparisons of South Africa to other locations, five outlier loci were identified: Orla2-91, Orla12-160, Orla6-313, Orla8-113 and Orla9-204 (Supplementary Table S5). No outlier loci were identified when considering only sampling sites within South Africa, or between the sites north of the upwelling zone.
Both the sequence and microsatellite loci indicated population subdivision between South Africa and the other locations. On the basis of cyt b, all pairwise comparisons involving South Africa were significant (average ΦST 0.61, P-value <0.001). When South Africa was excluded, pairwise tests showed no significant differences (average ΦST 0.02, P-value >0.05; Table 2). For AM2B1, all pairwise comparisons were significant, except between Senegal and Portugal (Table 2). Pairwise FST values between South Africa and the other sampling sites ranged from 0.15 to 0.20 for the microsatellite loci. Outlier microsatellite loci showed FST estimates ranging from 0.30 to 0.33 (P-value <0.001; Table 2), while for the non-outlier microsatellite loci, the FST was ~0.06 (P-value <0.001; Table 2). Among the sites north of the upwelling zone all pairwise differences were significant based on the combined loci, but none were significant when only outlier loci were considered (Table 2 and Supplementary Figure S2). This differing pattern could be due to the difference in marker numbers and number of alleles between the outlier and putatively neutral loci and should be considered when interpreting the patterns of differentiation (Supplementary Figure S2). All pairwise differences between sites within South Africa were not significant (Supplementary Table S4). The Bayesian clustering method identified South Africa as distinct from the other sites (ad hoc statistic ΔK= 2; Figures 1d and e).
Using the IMa model to estimate demographic parameters from the data, differing patterns and intensities of gene flow were identified between South Africa and the other East Atlantic locations (Table 3; Supplementary Figure S3). Analyses for the combined data set (outputs available in Supplementary Data S2) indicated unidirectional gene flow from South Africa to the East Atlantic (refer to Supplementary Table S6; Table 3 and Figure 2). Different marker types indicated varying patterns of gene flow. Similar patterns were observed with the outlier microsatellite loci and cyt b indicating unidirectional gene flow. The combined microsatellite loci indicated bidirectional gene flow in contrast to intron AM2B1 which indicated no migration (Supplementary Table S6; Supplementary Figure S3). Using the rate of molecular evolution of cyt b calculated for three-spined stickleback, the combined data set (cyt b, AM2B1 and all microsatellites) indicated a date of divergence of 220 kyr (90% HPD 77–395 kyr). This was inflated compared with the estimated divergence using only cyt b or the non-outlier microsatellite loci (~12–30 kyr). However, this coincided with the larger divergence found with the outlier microsatellite loci (200 kyr, HPD 110–280 kyr). All nuclear loci and combined loci indicated a much larger effective population size for the ancestral population. In all cases, the East Atlantic had a larger effective population size than South Africa (Supplementary Figure S3).

Calibrated posterior probability estimates for (a) divergence time in thousands of years (b) effective population size in thousands of individuals (NE) and (c) the extent and direction of migration (in number of migrations per 1000 of generations per gene copy) across the Benguela upwelling zone estimated in IMa 1.0. Three individual runs (with differing random seeds) are indicated by varying shades of color and are coded as follows: (a) divergence time in shades of black to grey (b) NE for South Africa in greens, NE for East Atlantic in oranges and NE for ancestral populations in greys (c) migration into South Africa in greens and migration into East Atlantic in oranges.
Discussion
Genetic structure and connectivity of P. saltatrix along the western coast of Africa
The main findings of this study identified a divergence between South Africa and the rest of the East Atlantic (Portugal, Senegal and Angola) with recent asymmetrical secondary contact. The molecular markers used showed varying patterns of divergence and introgression with the nuclear loci (microsatellites and intron) showing a clear genetic break and the mtDNA current yet limited gene flow. In addition, no structure was found across biogeographic provinces of South Africa and only weak subdivision was identified in the East Atlantic from Angola to Portugal.
The major genetic pattern indicated a divergence between South Africa and the rest of the East Atlantic. The divergence estimates and the presence of gene flow identified through IMa (Figure 2 and Table 3) indicated a period of isolation of the South African population with recent secondary contact in Angola. Several individuals from Angola shared cyt b haplotypes with South Africa. Through the sequencing of these individuals for additional mtDNA regions (CR and ND2) it was evident that ancestral polymorphism could not explain the observed patterns of shared haplotypes (Figure 1 and Supplementary Figure S1). These Angolan samples of South African origin did not have new specific mutations, indicating that this secondary contact is recent, unlike other instances of secondary contact events from South-East Atlantic/Indian Ocean into the North-east Atlantic (Lithognathus mormymus; Sala-Bozano et al., 2009).
Among the fish found further north (Angola, Senegal, Portugal), three haplogroups could be distinguished, although no geographical clustering was observed (Table 3 and Figure 1b). This could be the result of the stochastic coalescent process, but could also suggest past isolation followed by secondary contacts. In the latter case, this would be most likely due to the isolation during glacial maxima as observed in many Atlantic marine species (Maggs et al., 2008; Miralles et al., 2014b). The genetic sub-structuring identified among the sites north of Lüderitz (FST; and Figures 2b and c) has been observed in other species with a similar distribution to P. saltatrix (Durand et al., 2005b; Sala-Bozano et al., 2009; Durand et al., 2013), and has been attributed to the influence of glacial refugia (Miralles et al., 2014a) and species life-histories. During glacial periods, a number of species moved southwards from the North-east Atlantic due to the lack of suitable habitats, colder sea-surface temperatures and the reduction of connection between the North Atlantic and Mediterranean Sea (Maggs et al., 2008; Miralles et al., 2014b; Silva et al., 2014a). These results indicate that some southern locations are likely past refugia for marine species (Durand et al., 2005b). This might be the case for Angola, as it harbors a large amount of genetic diversity in all markers considered (Figures 1b and c; Table 1; Supplementary Table 3); this should be further explored.
The South African population that extends across two distinct biogeographic regions did not show any evidence to support divergence across the Agulhas–Benguela transition (Hedger et al., 2010; Teske et al., 2011). The F-statistics and the Bayesian clustering analyses supported a single population. This is in contrast to the previous results that found levels of residency in a coastal embayment (Hedger et al., 2010), indicating the possibility of two spawning locations, one on the west coast and one on the south coast of South Africa. The lack of genetic subdivision across the biogeographic barrier may be explained by the annual spawning migration of the species to coastal sites in the sub-tropical region of South Africa (Figure 1; Van der Elst, 1976). Moreover, it is well established that only a small amount of gene flow between separate populations (especially in marine species with large population sizes) could mask the genetic signal of distinct stocks (Hauser and Carvalho, 2008). Finally, P. saltatrix could have contrasting life-history strategies along the coasts of South Africa with only a portion of the population undertaking a spawning migration and another portion being residential (Hedger et al., 2010). This would again challenge the detection of distinct stocks. Although this phylogeographic transition has been described in a number of coastal inshore species (reviewed Teske et al., 2011, 2014), it is not a consistent barrier even for marine fish species with residential adults. These results indicate that the transport of pelagic larvae across this barrier is possible (Teske et al., 2010, 2011) and that P. saltatrix is a good example of such a species with a single genetic stock around South Africa.
The Benguela current upwelling zone as a barrier to gene flow
The strong divergence of P. saltatrix between South Africa and the rest of the East Atlantic can be explained by the Benguela upwelling region, which appears to be the main generator of divergence in the region when considering other marine species. Studies have shown that this upwelling zone acts as a barrier to gene flow in Lichia amia and Atractoscion aequidens (Henriques et al., 2012, 2014) and unique lineages/haplogroups to South Africa were observed in numerous species (for example, Membranipora membranacea and Lithognatus mormyrus; Schwaninger, 2008; Sala-Bozano et al., 2009; Teske et al., 2011). It is therefore likely that the Benguela upwelling zone is mostly impermeable, especially for shallow water species as it creates an unstable environment that restricts movement of larvae. However, limited bi-directional gene flow across the barrier has been observed in Agyrosomus inodorus, which suggests that the barrier can be partially permeable (Henriques et al., 2015), corroborating the results of a previous tagging study in Namibia (Kirchner and Holtzhausen, 2001).
On the basis of the known thermal range of eggs (18–22 °C; Norcross et al., 1974), early-life stages (17–24 °C) and adults (12–30 °C, Fahay et al., 1999) of P. saltatrix, the cold water (range 11–15 °C, Hardman-Mountford et al., 2003) associated with the large upwelling zone around Lüderitz is probably the main environmental barrier limiting gene flow. Therefore, movement across the upwelling zone is more likely to be infrequent and achieved by migrating adults during environmental anomalies, or could be a historical colonization during glacial periods (Miralles et al., 2014b). However, as the mtDNA suggests that the secondary contact is relatively recent, it is possible that secondary contact may occur during the Benguela El Niño events, which are characterized by pole-ward intrusions of warm water from the Angolan current (Florenchie et al., 2003). In addition, due to the upper thermal tolerance of the eggs and larvae (Norcross et al., 1974), it is unlikely that they could readily traverse the tropical barrier. If this is the case, the extent of connectivity observed among the locations north of the upwelling zone thus either reflects a signature of a recent secondary contact with neutral regions introgressed, or continuous events of adult migration between locations.
Ecological studies have shown that P. saltatrix is characterized by contrasting life-history traits between different locations across the East Atlantic and around South Africa. These may contribute to the observed patterns and directionality of secondary contact. The spawning period varies latitudinally, with populations in South Africa and Angola spawning September–March, in Senegal May–July and October–November and in the Western Mediterranean around July–August (Juanes et al., 1996). The nursery and spawning habitats also vary between regions (Juanes et al., 1996; Bealey, 2014). A large portion of the South African population undertakes an extensive migration annually to suitable spawning habitat, whereas in Angola the narrow distribution range of the population provides adequate temperatures for spawning during November–December and no spawning migration has been observed (WM Potts, personal observation).
Although this species shows variations in life-history characteristics within the study area, none of these can fully explain the genetic patterns observed, that is, the deep divergence of South Africa and the high connectivity among the locations further north of the upwelling zone. However, these variations in life-history traits likely also affect the ability for isolated populations to reconnect in this region.
Genetic heterogeneity across the genome of P. saltatrix
The presented data showed a range of genetic divergences, from moderate divergences observed across this barrier to uncharacteristically high divergence for marine fish at five outlier loci (FST ~0.30 P<0.001; Table 2, Figure 2), as well as at a nuclear intron AM2B1 (FST ~0.5 P<0.001). Such contrasting levels of divergence across the genome in P. saltatrix may be due to endogenous barriers leading to lack of introgression after secondary contact caused by Dobzhansky-Muller incompatibilities (Bierne et al., 2011).
The high proportion of outliers observed in our data (38%) and the clear evidence of a recent secondary contact across a semi-permeable oceanographic barrier indicates a plausible scenario of genome incompatibilities leading to variation in nuclear divergence patterns in P. saltatrix. Adaptive divergence would not lead to this high percentage of outliers as this process is normally observed at only a small fraction of the genome (0.002%; Meirmans, 2015). This high number of outliers could likely be due to the location of the microsatellite loci in intergenic regions, which are likely introgressing at a slower pace than random regions of the genome in the presence of genomic incompatibilities. The mtDNA indicates a larger introgression than the nuclear loci, as 15% of the sequenced cyt b from Angola (which was only 26 samples) had South African mtDNA haplotypes, indicating that there was likely a limitation on nuclear DNA introgression. Also, if introgression was recent and inconsistent mixing after secondary contact occurred (as is likely the case of P. saltatrix) only small portions of the South African genomic DNA would be present in the Angolan P. saltatrix and these signals could be maintained for many generations (Bierne et al., 2011, 2013).
The mtDNA divergence time points to a recent colonization and establishment of the South African population from the East Atlantic as indicated by IMa based on the calibration of demographic transition mutation rate (Table 3). However, portions of the nuclear genome appear to indicate much older divergence with almost no evidence of nuclear introgression, especially for the microsatellite outlier loci and the nuclear intron (Table 3 and Figures 1c and 2). When divergence occurs across a natural barrier we would expect a more uniform signal across the genome, as suggested by Bierne et al. (2011). After secondary contact in such a scenario, the neutral regions of the genome will be able to freely introgress. This is not the case in P. saltatrix where the main evidence for secondary contact is identified through mitochondrial introgression with almost no nuclear introgression. In addition, even though the upwelling zone has been identified to be a barrier to gene flow in several cases, it does not restrict all species movement in the region. Species with similar life-history characteristics to P. saltatrix (that is, adult dispersal potential) are more likely to cross the barrier sporadically than species with highly sedentary adults. This natural barrier indicates a phylogenetic break in many marine species but this upwelling zone is not always persistent and its strength fluctuates during seasons and climatic anomalies (Florenchie et al., 2003; Hutchings et al., 2009). Therefore, this explains the genetic breaks in several species, but not necessarily the maintenance of genetic differentiation in such a marine habitat.
Conclusions
Understanding why certain genomic regions are unable to introgress (either due to exogenous or endogenous barriers) provides insight into the evolution of species. Here we have shown a likely recent secondary contact event between the South African and Angolan populations of P. saltatrix, with contrasted nuclear introgression between genomic regions. Also, a deep divergence was observed across a limited geographic distance (1500 km), while genetic homogeneity was observed across a large region of the East Atlantic from Angola to Portugal and along the South African coast. Future research across this region should focus on several species with continuous sampling to better understand the role this barrier has in the maintenance of divergence of species distributed on either side. In addition, these contrasted signals within P. saltatrix will be better understood through comparisons with the other populations distributed globally and the implementation of genomics approaches.
Data archiving
Mitochondrial sequences available from GenBank: KX196669-KX196754. Microsatellite data and IMa files available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.11cd0.
Accession codes
References
Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart G . (2008). LOSITAN: a workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinformatics 9: 323.
Barton NH . (1979). The dynamics of hybrid zone. Heredity 43: 341–359.
Bealey RSJ (2014). Short term changes to the life history of shad, Pomatomus saltatrix (Perciformes: Pomatomidae), in southern Angola. MSc thesis, Rhodes University.
Bierne N, Welch J, Loire E, Bonhomme F, David P . (2011). The coupling hypothesis: why genome scans may fail to map local adaptation genes. Mol Ecol 20: 2044–2072.
Bierne N, Gagnaire PA, David P . (2013). The geography of introgression in a patchy environment and the thorn in the side of ecological speciation. Curr Zool 59: 72–86.
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F . (1996–2004) GENETIX 4.05 Logiciel sous Windows pour la Génétique des Populations, Laboratoire Génome, Populations, Interactions, CNRS UMR 5171. Université de Montpellier II: Montpellier, France.
Chapuis MP, Estoup A . (2007). Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24: 621–631.
Chen YZ, Xu TJ, Jin XX, Tang D, Wei T, Sun YY et al. (2012). Universal primers for amplification of the complete mitochondrial control region in marine fish species. Mol Biol 46: 727–730.
Chikhi L, Bonhomme F, Agnese JF . (1998). Low genetic variability in a widely distributed and abundant clupeid species, Sardinella aurita. New empirical results and interpretations. J Fish Biol 52: 861–878.
Clement M, Posada D, Crandall KA . (2000). TCS: a computer program to estimate gene genealogies. Mol Ecol 9: 1657–1659.
Cowen RK, Lwiza KMM, Sponaugle S, Paris CB, Olson DB . (2000). Connectivity of marine populations: open or closed? Science 287: 857–859.
Dos Santos SMR, Klopper AW, Oosthuizen CJ, Bloomer P . (2008). Isolation and characterization of polymorphic tetranucleotide microsatellite loci in the pelagic perciform fish Pomatomus saltatrix (Linnaeus, 1766) from South Africa. Mol Ecol Res 8: 1065–1067.
Durand JD, Collet A, Chow S, Guinand B, Borsa P . (2005a). Nuclear and mitochondrial DNA markers indicate unidirectional gene flow of Indo-Pacific to Atlantic bigeye tuna (Thunnus obesus populations, and their admixture off southern Africa. Mar Biol 147: 313–322.
Durand JD, Tine M, Panfili J, Thiaw OT, Lae R . (2005b). Impact of glaciations and geographic distance on the genetic structure of a tropical estuarine fish, Ethmalosa fimbriata (Clupeidae, S. Bowdich,1825). Mol Phylogenet Evol 36: 277–287.
Durand JD, Guinand B, Dodson JJ, Lecomte F . (2013). Pelagic life and depth: coastal physical features in West Africa shape the genetic structure of the Bonga Shad, Ethmalosa fimbriata. PloS One 8: e77483.
Earl DA, VonHoldt BM . (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Con Gen Res 4: 359–361.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: as simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Laval G, Schneider S . (2005). Arlequin version 3.11: an integrated software package for population genetics data analysis. Evol Bioinform Online 1: 47–50.
Fahay MP, Berrien PL, Johnson DL, Morse WW . (1999) Essential Fish Habitat Source Document: Bluefish, Pomatomus saltatrix, Life History and Habitat Characteristics NOAA Technical Memo. NMFS-NE-144, 68 p National Marine Fisheries Service: Woods Hole, MA, USA.
Florenchie P, Lutjeharms JRE, Reason CJC, Masson S, Rouault M . (2003). The source of Benguela Niños in the South Atlantic Ocean. Geophys Res Lett 30: 1505.
Gagnaire P-A, Minegishi Y, Aoyama J, R'eveillac E, Robinet T, Bosc P et al. (2009). Ocean currents drive secondary contact between Anguilla marmorata populations in the Indian Ocean. Mar Ecol Prog Ser 379: 267–278.
Gagnaire PA, Minegishi Y, Zenboudji S, Valade P, Aoyama J, Berrebi P . (2011). Within-population structure highlighted by differential introgression across semipermeable barriers to gene flow in Anguilla marmorata. Evolution 65: 3513–3525.
Gagnaire PA, Broquet T, Aurelle D, Viard F, Souissi A, Bonhomme F . (2015). Using neutral, selected, and hitchhiker loci to assess connectivity of marine populations in the genomic era. Evol Appl 8: 769–786.
Gaither MR, Bowen BW, Bordenave TR, Rocha LA, Newman SJ, Gomez JA et al. (2011). Phylogeography of the reef fish Cephalopholis argus (Epinephelidae) indicates Pleistocene isolation across the indo-pacific barrier with contemporary overlap in the coral triangle. BMC Evol Biol 11: 189.
Goodbred CO, Graves JE . (1996). Genetic relationships among geographically isolated populations of bluefish (Pomatomus saltatrix. Mar Fresh Res 47: 347–355.
Gotoh RO, Tamate S, Yokoyama J, Tamate HB, Hanzawa N . (2013). Characterization of comparative genome‐derived simple sequence repeats for acanthopterygian fishes. Mol Ecol Res 13: 461–472.
Grant WS, Bowen BW . (1998). Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation. J Hered 89: 415–426.
Hardman-Mountford NJ, Richardson AJ, Agenbag JJ, Hagen E, Nykjaer L, Shillington FA et al. (2003). Ocean climate of the South East Atlantic observed from satellite data and wind models. Prog Oceanogr 59: 181–221.
Hassan M, Lemaire C, Fauvelot C, Bonhomme F . (2002). Seventeen new exon‐primed intron‐crossing polymerase chain reaction amplifiable introns in fish. Mol Ecol Not 2: 334–340.
Hauser L, Carvalho GR . (2008). Paradigm shifts in marine fisheries genetics: ugly hypotheses slain by beautiful facts. Fish Fish 9: 333–362.
Hedger RD, Næsje TF, Cowley PD, Thorstad EB, Attwood C, Økland F et al. (2010). Residency and migratory behaviour by adult Pomatomus saltatrix in a South African coastal embayment. Estuar Coast Shelf Sci 89: 12–20.
Hellberg ME . (2009). Gene flow and Isolation among populations of marine animals. Annu Rev Ecol Evol Syst 40: 291–310.
Hemmer-Hansen J, Nielsen EE, Grønkjær P, Loeschecke V . (2007). Evolutionary mechanisms shaping the genetic population structure of marine fishes; lessons from the European flounder (Platichthys flesus L.). Mol Ecol 16: 3104–3118.
Henriques R, Potts WM, Sauer WHH, Shaw PW . (2012). Evidence of deep genetic divergence between populations of an important recreational fishery species, Lichia amia L. 1758, around southern Africa. Afri J Mar Sci 34: 585–591.
Henriques R, Potts WM, Santos CV, Sauer WHH, Shaw PW . (2014). Population connectivity and phylogeography of a coastal fish, Atractoscion aequidens (Sciaenidae), across the Benguela current region: evidence of an ancient vicariant event. PLoS One 9: e87907.
Henriques R, Potts WM, Sauer WHH, Shaw PW . (2015). Incipient genetic isolation of a temperate migratory coastal sciaenid fish (Argyrosomus inodorus within the Benguela cold current system. Mar Biol Res 11: 423–429.
Hewitt GM . (1988). Hybrid zones—natural laboratories for evolutionary studies. Trends Ecol Evol 3: 158–167.
Hey J, Nielsen R . (2007). Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc Natl Acad Sci USA 104: 2785–2790.
Ho SYW, Phillips MJ, Cooper A, Drummond AJ . (2005). Time dependency of molecular rate estimates and systematic overestimation of recent divergence times. Mol Biol Evol 22: 1561–1568.
Hoareau TB, Boissin E . (2010). Design of phylum‐specific hybrid primers for DNA barcoding: addressing the need for efficient COI amplification in the Echinodermata. Mol Ecol Res 10: 960–967.
Hoareau TB . (2016). Late-glacial demographic expansion motivates a clock overhaul for population genetics. Syst Biol 65: 449–464.
Hubisz MJ, Falush D, Stephens M, Pritchard JK . (2009). Inferring weak population structure with the assistance of sample group information. Mol Ecol Res 9: 1322–1332.
Hutchings L, van der Lingen CD, Shannon LJ, Crawford RJM, Verheye HMS, Bartholomae CH et al. (2009). The Benguela current: an ecosystem of four components. Prog Oceanogr 83: 15–32.
Juanes F, Hare JA, Miskiewicz AG . (1996). Comparing early life history strategies of Pomatomus saltatrix: a global approach. Mar Fresh Res 47: 365–379.
Kimura M, Ohta T . (1978). Stepwise mutation model and distribution of allelic frequencies in a finite population. Proc Natl Acad Sci USA 75: 2868–2872.
Kirchner CH, Holtzhausen JA . (2001). Seasonal movements of silver kob, Argyrosomus inodorus, (Griffiths and Heemstra) in Namibian waters. Fish Manag Ecol 8: 239–251.
Larmuseau MHD, Raeymaekers JAM, Ruddick KG, van Houdt JKJ, Volckaert FAM . (2009). To see in different seas: spatial variation in the rhodopsin gene of the sand goby (Pomatoschistus minutus. Mol Ecol 18: 4227–4239.
Librado P, Rozas J . (2009). DnaSP v5: as software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Maggs CA, Castilho R, Foltz D, Henzler C, Jolly NT, Kelly J et al. (2008). Evaluating signatures of glacial refugia for North Atlantic benthic marine taxa. Ecology 89: 108–122.
Meirmans G . (2015). Seven common mistakes in population genetics and how to avoid them. Mol Ecol 24: 3223–3231.
Miralles L, Juanes F, Garcia-Vazquez E . (2014a). Interoceanic sex-biased migration in bluefish. Trans Am Fish Soc 143: 1308–1315.
Miralles L, Juanes F, Pardiñas AF, Garcia-Vazquez E . (2014b). Paleoclimate shaped bluefish structure in the northern hemisphere. Fisheries 39: 578–586.
Norcross JJ, Richardson SL, Massmann WH, Joseph EB . (1974). Development of young bluefish (Pomatomus saltatrix and distribution of eggs and young in Virginian coastal waters. Trans Am Fish Soc 103: 477–497.
O’Reilly PTO, Canino MF, Bailey KM, Bentzen P . (2004). Inverse relationship between FST and microsatellite polymorphism in the marine fish, walleye Pollock (Theragra chalcogramma: implications for resolving weak population structure. Mol Ecol 13: 1799–1814.
Palumbi SR . (1994). Genetic divergence, reproductive isolation and marine speciation. Annu Rev Ecol Syst 25: 547–572.
Pardiñas AF, Campo D, Pola IG, Miralles L, Juanes F, Garcia‐Vazquez E . (2010). Climate change and oceanic barriers: genetic differentiation in Pomatomus saltatrix (Pisces: Pomatomidae) in the North Atlantic Ocean and the Mediterranean Sea. J Fish Biol 77: 1993–1998.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Quéré N, Desmarias E, Tsigenopoulos CS, Belkhir K, Bonhomme F, Guinand B . (2012). Gene flow at major transitional areas in sea bass (Dicentrarchus labrax and the possible emergence of a hybrid swarm. Ecol Evol 2: 3061–3078.
Rice WR . (1989). Analyzing tables of statistical tests. Evolution 43: 223–225.
Rousset F . (2008). Genepop'007: a complete reimplementation of Genepop software for Windows and Linux. Mol Ecol Res 8: 103–106.
Roux C, Tsagkogeorga G, Bierne N, Galtier N . (2013). Crossing the species barrier: genomic hotspots of introgression between two highly divergent Ciona intestinalis species. Mol Biol Evol 30: 1574–1578.
Schwaninger HR . (2008). Global mitochondrial DNA phylogeography and biogeographic history of the antitropically and longitudinally disjunct marine bryozoan Membranipora membranacea L. (Cheilostomata): Another cryptic sibling species complex? Mol Phylogenet Evol 49: 893–908.
Sebastés A, Martín P, Raya V . (2012). Changes in life-history traits in relation to climate change: bluefish (Pomatomus saltatrix in the northwestern Mediterranean. ICES J Mar Sci 69: 1000–1009.
Sala-Bozano M, Ketmaier V, Mariani S . (2009). Contrasting signals from multiple markers illuminate population connectivity in a marine fish. Mol Ecol 18: 4811–4826.
Selkoe KA, Toonen RJ . (2006). Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol Lett 9: 615–629.
Silva G, Horne JB, Castilho R . (2014a). Anchovies go north and west without losing diversity: post-glacial range expansions in a small pelagic fish. J Biogeogr 41: 1171–1182.
Silva G, Lima FP, Martel P, Castilho R . (2014b). Thermal adaptation and clinal mitochondrial DNA variation of European anchovy. Proc R Soc Lond B Biol Sci 281: 20141093.
Stephens M, Smith NJ, Donnelly P . (2001). A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68: 978–989.
Stephens M, Donnelly P . (2003). A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 73: 1162–1169.
Tajima F . (1983). Evolutionary relationship of DNA sequences in finite populations. Genetics 105: 437–460.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S . (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739.
Teske PR, Forget FRG, Cowley PD, von der Heyden S, Beheregaray LB . (2010). Connectivity between marine reserves and exploited areas in the philopatric reef fish Chrysoblephus laticeps (Teleostei: Sparidae). Mar Biol 157: 2029–2042.
Teske PR, von der Heyden S, McQuaid CD, Barker NP . (2011). A review of marine phylogeography in southern Africa. S Afr J Sci 107: 43–53.
Teske PR, Papadopoulos I, Barker NP, McQuaid CD, Beheregaray LB . (2014). Mitonuclear discordance in genetic structure across the Atlantic/Indian Ocean biogeographical transition zone. J Biogeogr 41: 392–401.
Thompson JD, Higgins DG, Gibson TJ . (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680.
Van der Elst R. (1976). Game Fish of the East Coast of South Africa. I. The Biology of the Elf, Pomatomus saltatrix (Linnaeus), in the Coastal Waters of Natal. Oceanographic Research Institute. South African Association for Marine Biological Research Investigational Report 44.
Watterson GA . (1975). On the number of segregating sites in genetical models without recombination. Theor Popul Biol 7: 256–276.
Weir BS, Cockerham CC . (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Zane L, Bargelloni L, Patarnello T . (2002). Strategies for microsatellite isolation: a review. Mol Ecol 11: 1–16.
Acknowledgements
We acknowledge the African Coelacanth Ecosystem Programme (ACEPII) and the International Foundation of Science (IFS, grant A_5216_1) for financial and logistical support. This work is based on research supported by the National Research Foundation (NRF) of South Africa and IFS; any opinion, finding and conclusion expressed in this material are that of the authors and the NRF and IFS does not accept liability in regard thereto. We would like to acknowledge Patrice Brehmer, Omar Ndiaye, Mor Sylla and the AWA consortium as we as Chantal Conand, Lorraine Harris, Carel Oosthuizen, Evert Klopper, Greg Brett and Ocean and Coasts, Department of Environmental Affairs (South Africa) for facilitating sampling. We would also like to thank the DNA Sequencing Facility of the Faculty of Natural and Agricultural Sciences at the University of Pretoria and specifically acknowledge the use of the ABI3500xl.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Rights and permissions
About this article

Cite this article
Reid, K., Hoareau, T., Graves, J. et al. Secondary contact and asymmetrical gene flow in a cosmopolitan marine fish across the Benguela upwelling zone. Heredity 117, 307–315 (2016). https://doi.org/10.1038/hdy.2016.51
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2016.51
This article is cited by
-
Spatial and temporal stability in the genetic structure of a marine crab despite a biogeographic break
Scientific Reports (2022)
-
Three mitochondrial lineages and no Atlantic-Mediterranean barrier for the bogue Boops boops across its widespread distribution
Scientific Reports (2022)
-
Global phylogeography of sailfish: deep evolutionary lineages with implications for fisheries management
Hydrobiologia (2021)
-
Mechanisms of peripheral phylogeographic divergence in the indo-Pacific: lessons from the spiny lobster Panulirus homarus
BMC Evolutionary Biology (2017)
