
Abstract
Understanding the genetic structure of a population is essential to its conservation and management. We report the level of genetic diversity and determine the population structure of a cryptic deep ocean cetacean, the Gray’s beaked whale (Mesoplodon grayi). We analysed 530 bp of mitochondrial control region and 12 microsatellite loci from 94 individuals stranded around New Zealand and Australia. The samples cover a large area of the species distribution (~6000 km) and were collected over a 22-year period. We show high genetic diversity (h=0.933–0.987, π=0.763–0.996% and Rs=4.22–4.37, He=0.624–0.675), and, in contrast to other cetaceans, we found a complete lack of genetic structure in both maternally and biparentally inherited markers. The oceanic habitats around New Zealand are diverse with extremely deep waters, seamounts and submarine canyons that are suitable for Gray’s beaked whales and their prey. We propose that the abundance of this rich habitat has promoted genetic homogeneity in this species. Furthermore, it has been suggested that the lack of beaked whale sightings is the result of their low abundance, but this is in contrast to our estimates of female effective population size based on mitochondrial data. In conclusion, the high diversity and lack of genetic structure can be explained by a historically large population size, in combination with no known exploitation, few apparent behavioural barriers and abundant habitat.
Similar content being viewed by others
Introduction
Population history, demography and behaviour all interact to shape the genetic diversity of a species. Species with fragmented or reduced populations often have low levels of genetic diversity. In contrast, high genetic diversity is consistent with long-term stability in population size, whereas low levels of population differentiation suggest connectivity or recent population expansion. As a rule, cetaceans are known to have low genetic diversity in comparison with terrestrial mammals and this is hypothesized to be due to slow mutation rates (Jackson et al., 2009) and demographic factors such as recent population expansion following bottlenecks or behaviour (Oremus et al., 2009).
Patterns of population structure are evident in most cetacean species, even those with seemingly continuous distributions and high mobility. For example, many baleen whales are highly philopatric, returning to calving or feeding grounds each year, leading to patterns of population structure between these grounds (Alter et al., 2009). The long lifespan of these animals, and extended period of maternal care, allows the cultural transmission of this philopatry over long time periods despite significant population depletion. In killer whales (Orcinus orca), population differentiation is thought to result from a highly matrifocal social system (Hoelzel et al., 1998). It has been suggested that the lack of gene flow between such matrifocal groups has, in the longer term, led to the development of sympatric subspecies that are specific to a particular habitat or prey (Morin et al., 2010). Furthermore, some wide-ranging cetacean species show local specialization. For example, common dolphins (Delphinus delphis) are a highly mobile pelagic species, yet genetic differentiation has been detected between animals from South Australia and those from the eastern coast of Tasmania, ~1500 km apart (Bilgmann et al., 2008). This differentiation is likely due to a dependence on specific regional oceanographic features such as upwellings that influence prey distributions, for example, the Bonney Upwelling (Butler et al., 2002). Whether these trends are evident in beaked whales has previously been unknown, and difficult to quantify given the problem with obtaining genetic samples.
Although patterns of population structure are known in other cetaceans, beaked whales remain enigmatic, with few published studies describing their populations. The ziphiids, or beaked whales, are one of the most speciose families of cetaceans, second only to the delphinids. Of the 22 species of beaked whale, 15 can be found within the genus Mesoplodon. Members of this genus are cryptic in their appearance and behaviour (Pitman, 2009). It has been assumed that their general biology is similar and most are thought to be deep-diving squid eaters that live in small groups along continental shelf edges. However, much of the information on the biology of these whales is derived from stranded animals combined with extrapolation from data collected on the few species that can be observed at sea (e.g., Wimmer and Whitehead, 2004; McSweeney et al., 2007).
Long-term cetacean sighting surveys off the east coast of the United States suggest that beaked whales cluster into ecological niches that are different from all other odontocetes (Schick et al., 2011). For example, Cuvier’s (Ziphius cavirostris) and Sowerby’s (Mesoplodon bidens) beaked whales occupy a different area of the continental shelf than do other squid-eating species such as sperm whales (Physeter macrocephalus). Moreover, there is evidence that these two beaked whale species may, in turn, occupy slightly different habitats within this area. Tagging data from Blainville’s beaked whales (Mesoplodon densirostris) have revealed a relationship between foraging behaviour and oceanographic features, for example, water depth, movements of the deep scattering layer of mesopelagic prey and seabed topography (Johnson et al., 2008). Indeed, tagging data suggest that Cuvier’s beaked whales perform the deepest dives of all mammals, almost 3000 m (Schorr et al., 2014). Furthermore, modelling implies that beaked whales require larger, higher quality habitats than other cetaceans to meet the energetic needs of such deep diving (Wright et al., 2011; New et al., 2013).
Where surveys have facilitated population size estimates, for example, west coast United States, it has been found that many beaked whale species have undergone significant population declines (Moore and Barlow, 2013). As beaked whales are particularly vulnerable to anthropogenic noise, it is speculated that these population declines are because of an increase in anthropogenic disturbance, further highlighting the need for a better understanding of the basic spatial requirements of all beaked whale species globally (Weilgart, 2007). However, in most areas of the world, and for most beaked whale species, these data are not available. The status of species inhabiting the remote areas of the Southern Ocean and seas around New Zealand is, as yet, undetermined.
New Zealand has the highest recorded number of species of stranded beaked whale in the world; 13 of the 22 recognized species, and some of the most rarely sighted (Thompson et al., 2012; Thompson et al., 2013; Constantine et al., 2014). One of the most frequent species to strand around the coast is the Gray’s beaked whale (Mesoplodon grayi) (Figure 1). This whale is a medium-sized (4.0–5.5 m) mesoplodont with a circumpolar southern hemisphere distribution (Figure 2a). Distributions have been primarily inferred from analyses of stranding data and live sightings are extremely rare. Most records are generally from south of 33° latitude, particularly on New Zealand, Australian, South African and South American coasts, including the sub-Antarctic and Antarctic waters, with one record from the Netherlands (Boschema, 1950; Dalebout et al., 2004; Taylor et al., 2008; Van Waerebeek et al., 2010; Scheidat et al., 2011). Similar to other beaked whales, Gray’s are assumed to live along the continental shelf edge, although there are occasional sightings of animals in shallow waters (e.g., Dalebout et al., 2004). An analysis of stranding patterns around New Zealand suggests that summer peaks are associated with inshore movements related to calving or nursing, particularly around the North Island (Thompson et al., 2013). Moreover, Gray’s beaked whales have subtle morphological differences between the east and west coasts of New Zealand (Thompson et al., 2014) and this might indicate restricted gene flow between the two coasts.

A stranded male Gray's beaked whale (M. grayi) at Pataua beach, in the North East region of New Zealand in December 2009.

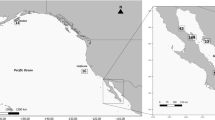
(a) The likely global distribution of Gray's beaked whales (M. grayi) based on both sightings and stranding records (follows International Union for Conservation of Nature listing, www.redlist.org). (b and c) Location of Gray’s beaked whale samples and a priori geographic regions. Stranding locations for samples are shown by black circles and sample numbers are given within parentheses. A priori regions are shown by colour (West Australia (purple), in New Zealand, North East (blue), North West (green), South East (orange) and South West (yellow)). For details of actual stranding locations, see Supplementary Information.
Behavioural studies of other beaked whales, for example, the northern bottlenose whale (Hyperoodon ampullatus), suggest specific dependencies on oceanographic features, such as submarine canyons, have resulted in genetically isolated populations (Dalebout et al., 2006). To date, there is no information on the foraging habitat or prey of Gray’s beaked whales, although MacLeod et al. (2003) have speculated that this species relies more on small benthic fish than other beaked whales. The seabed topography around New Zealand is diverse, supporting a variety of mesopelagic squid and fish (De Leo et al., 2010). Around the continental shelf edge, there are several areas of periodic high primary productivity resulting from upwelling of slope-associated deep water (MacDiarmid et al., 2013). Many marine mammals are known to take advantage of these upwellings for foraging (Torres, 2013; Sagnol et al., 2014). In the case of Gray’s beaked whales, it is unclear whether the species take advantage of these upwellings, although stranding patterns appear to indicate use of the highly productive areas of the continental shelf of the north east of the North Island of New Zealand, particularly in summer (Thompson et al., 2013). We hypothesize that, given the spatial scale from the east to west coast of New Zealand, we would expect genetic structure in Gray’s beaked whales in line with morphological differences, with habitat dependency, as a result of specialization to local prey or breeding areas, as the driver of differentiation. Furthermore, we suggest that such genetic divergence is likely to be greater over a larger spatial scale (6000 km) between the Chatham Islands and Western Australia.
To test this hypothesis, we analysed sequence data from 530 bp of mitochondrial control region and 12 variable microsatellite loci to investigate diversity, population structure and effective population size from 94 Gray’s beaked whale samples. These samples were collected from strandings around the coast of New Zealand, with an additional six samples from Western Australia representing the largest global collection of this species. We provide novel insights into the population dynamics of this enigmatic, and rarely sighted, species.
Materials and methods
Study area and sample collection
We collected samples from a region spanning more than 6000 km extending from the west coast of Australia to the Chatham Islands (New Zealand) in the east (Figure 2). Samples from New Zealand were obtained from the New Zealand Cetacean Tissue Archive and cover the period from 1991 to 2013. Samples from Australia were collected over a 3-year period and were obtained from the Western Australian Museum (for specimen details see Supplementary Tables S1 and S2 and Supplementary Information). Further samples from South Australia and Tasmania were not available in sufficient numbers to allow any meaningful analyses. Sex of all samples was determined by amplification of the SRY gene multiplexed with a ZFX/ZFY-positive control (Aasen and Medrano, 1990; Gilson et al., 1998; Thompson et al., 2012). Samples from New Zealand were divided into four a priori regional areas according to where on the coast the animal was found and the Australian samples provided a fifth regional grouping (Figures 2b and c). These areas were based on the location of known marine biogeographic barriers resulting from seabed topography and oceanographic currents (Ayers and Waters, 2005).
DNA extraction, sequencing and genotyping
Genomic DNA was isolated from tissue using proteinase K digestion followed by a standard 25:24:1 Phenol:Chloroform:Isoamyl protocol as described by Sambrook et al. (1989) and modified by Baker et al. (1994), followed by ethanol precipitation. A 530 bp fragment of DNA from the mitochondrial control region was amplified and sequenced in both directions according to methods described in Thompson et al. (2013) using the primer pair Dlp1.5 and Dlp8G. Sequences were trimmed by eye in the programme GENEIOUS v7.1 (www.geneious.com), and only those sequences that reached a PHRED score of 40 or above for at least 70% of individual bases were deemed acceptable for analyses (Kearse et al., 2012). The first base of the control region was designated to be position 15 468 in reference to the Gray’s beaked whale whole mitogenome (GenBank accession no. KF981442). Sequences were aligned using MAFFT (Multiple Alignment using Fast Fourier Transform) multiple sequence alignment tool (Katoh et al., 2002).
Genotype data from 12 microsatellite loci (one di-, three tri- and eight tetra- repeats) were obtained using primers and methods developed by Patel et al. (2014). MICROCHECKER (Van Oosterhout et al., 2004) was used to assess evidence of scoring error due to stuttering, large allele dropout and null alleles. Deviation from Hardy–Weinberg equilibrium and linkage disequilibrium between loci were tested in ARLEQUIN 3.5 (Excoffier and Lischer, 2010).
Genetic diversity
For mitochondrial DNA (mtDNA) data, haplotypic (h) and nucleotide (π) diversities were calculated using ARLEQUIN. The nucleotide substitution model used to calculate genetic distance was the Tamura and Nei model with a gamma correction of α=0.219 as determined in jModelTest using the corrected Akaike information criterion (Tamura and Nei, 1993; Posada, 2008). For genotype data, average allelic richness (Rs), observed and expected heterozygosities were calculated per microsatellite locus and per putative population using GENODIVE 2.3b23 (Meirmans and Van Tienderen, 2004). Measures of genetic diversity can be highly dependent on sample size, in that larger populations are likely to have more alleles than smaller populations; therefore, allelic richness values were also calculated per population using the rarefaction method implemented in HP-RARE 1.0 (Kalinowski, 2005). This method statistically adjusts for sample size by calculating the number of alleles as a function of the sample size per population.
To identify any genetic signature of demographic expansion or population bottleneck in the mtDNA, Fu’s Fs statistic was calculated, as implemented in ARLEQUIN. Fu’s Fs is one of the more sensitive indicators of deviation from neutral population equilibrium (Ramos-Onsins and Rozas, 2002). Departure from neutral expectation was inferred by randomization using a coalescent algorithm run for 10 000 steps (Hudson, 1990). Negative values of Fu’s Fs statistic are indicative of historical population expansion or genetic hitchhiking, and a positive value is evidence of a recent population bottleneck and a deficiency of alleles at this locus (Fu, 1997).
Population structure
To visualize the geographic distribution of mtDNA haplotypes and their relationships, the program POPART was used to construct a median joining network (University of Otago, Dunedin, New Zealand; http://popart.otago.ac.nz). We estimated a phylogenetic tree of samples using a variant of Bayesian inference (Mr Bayes) with two Markov chain Monte Carlo sampler runs of 1.1 × 106 generations and the nucleotide substitution model as determined by jModelTest (Huelsenbeck and Ronquist, 2001). Blainville’s beaked whale (M. densirostris) and Gervais’ beaked whale (Mesoplodon europeaus) were selected as outgroups. Trees were sampled every 200 generations and it was determined by visual inspection of posterior traces of both runs that stationarity was attained by 1.1 × 105 generations. The first 1.1 × 105 generations were discarded as burn-in leaving the remaining samples to estimate a consensus tree and posterior probabilities.
An analysis of molecular variance and pairwise F-statistics were calculated for mtDNA and microsatellites in ARLEQUIN and GENODIVE, respectively. Three F-statistics were used: standard Fst based on mtDNA haplotype and microsatellite allele frequencies; Φst that incorporates molecular sequence divergence in mtDNA and, F′st that is a standardised Fst statistic that takes into account within-population genetic variation for microsatellites (Meirmans and Hedrick, 2011). Pairwise exact tests were also carried out and significance of all F-statistics was tested using 10 000 permutations. Analysis of molecular variance and F-statistics were analysed by each sex separately (data not shown) and for the combined data set.
Population structure was also investigated using a Bayesian clustering analysis to estimate the most probable number of populations using STRUCTURE 2.3.4 (Pritchard et al., 2000). Analysis of microsatellite data was conducted with and without sampling location priors using the admixture model. The number of clusters (K) with the highest posterior probability was identified using replicate runs assuming K from 1 to 5. The burn-in length was set at 100 000 steps, followed by 1 000 000 steps with a total of 10 replicates for each value of K. The most likely number of homogeneous clusters was assessed using the second-order rate of change or ΔK method following Evanno et al. (2005) and implemented in STRUCTURE HARVESTER (Earl and vonHoldt, 2012). Results were then combined in the program CLUMPP to average individual clustering outputs between runs (Jakobsson and Rosenberg, 2007) and visualized in DISTRUCT (Rosenberg, 2004). A principal component analysis was used to further visualize differences in genotypic variation between populations and individuals as implemented in R using the package ADEGENET (Jombart, 2008).
Estimating effective population size
Mitochondrial control region sequences were used to estimate effective female population size (Nef) using a Bayesian skyline plot approach implemented in BEAST v.1.8.0 (Drummond et al., 2012). The substitution model (TN93) with discrete gamma distribution with four rate categories was selected as the model of evolution having been previously determined in jModelTest. A strict molecular clock approach was used that assumed a control region mutation rate of 0.9 × 10−8 bp per year (Cuvier’s beaked whales; Dalebout et al., 2005), and 2 × 10−7 bp per year derived from ancient DNA sampling (bowhead whales (Balaena mysticetus); Ho et al., 2007, 2011). The Markov chain Monte Carlo chains were run with 3 × 107 iterations and samples were drawn every 30 000 iterations with the first 10% being discarded as burn-in. Population history was inferred using the Bayesian skyline plot with 10 groups of coalescent intervals. Two independent BEAST analyses were combined and in all cases convergence to stationary distribution and sufficient sampling were visually checked in TRACER v.1.6 (Rambaut et al., 2013).
Results
Genetic diversity
A total of 94 individuals were sequenced resulting in 38 mitochondrial haplotypes defined by 26 variable sites (Supplementary Table S3 and Supplementary Information; GenBank accession numbers: KJ767593–KJ767630). Diversity statistics suggest that Gray’s beaked whales have moderately high levels of variation within the study areas and both haplotype (h) and nucleotide (π) diversity were found to be similar between regions (Table 1). The same 94 individuals were genotyped at 12 microsatellite loci. The average number of alleles (k), allelic richness and private allelic richness were similar among regions. The only exception being k, which was lower in both the south west of New Zealand and Western Australia where there were fewer samples than in the other areas (Table 1). No microsatellite loci deviated from Hardy–Weinberg equilibrium and there was no significant linkage disequilibrium between loci after Bonferroni correction (Supplementary Table S4 and Supplementary Information). Loci showed no evidence for null alleles, large allelic dropout or scoring errors due to stutter peaks. The average amount of missing allelic data per locus was 0.35%. Diversity statistics per loci are given in Supplementary Table S5.
Fu’s Fs value was negative and highly significant (−23.01, P<0.001) and indicative of historical demographic expansion or selective sweep, and an excess of rare substitutions and haplotypes at this locus. These results suggest that it is unlikely that Gray’s beaked whales have suffered any historical genetic bottleneck.
Population structure
The median joining network of haplotypes showed no phylogeographic structure and common haplotypes were shared across the study area (Supplementary Figure S1). Moreover, the network is highly reticulated and most haplotypes are sister lineages in that they differ by only a single substitution. This pattern is also reflected in the Bayesian tree (Supplementary Figure S2).
Pairwise comparisons between populations showed no significant differentiation in either mtDNA or microsatellites at the P<0.05 level (Table 2). This lack of significance held true whether the sexes were combined or separated (data not shown). Bayesian clustering analyses implemented in STRUCTURE showed no population structure for microsatellite data. The highest average posterior probability occurred at K=1 and graphical outputs from DISTRUCT showed that with increasing values of K, all populations became increasingly subdivided into multiple clusters approximately proportional to the sample size for a priori regions (Figure 3). Both STRUCTURE analyses, with and without priors, revealed the same results. Principal component analysis showed all populations overlapping in genotypes with no visible differentiation between any of the a priori regions (Supplementary Figure S3).

Bayesian STRUCTURE analysis of 12 Gray’s beaked whale (M. grayi) microsatellite loci from five a priori regions. Each bar represents the likelihood of an individual's assignment to a particular population cluster as indicated by the colours for K=2–5.
Estimate of effective population size
Using the mutation rate derived from Cuvier’s beaked whale, the product of female Nef and generation time was calculated to be 10.14 million with 95% credibility intervals of 0.39–51.79 million. Using the faster mutation rate from bowhead whales, the product of female Nef and generation time was calculated to 0.46 million with 95% credibility intervals of 0.02–2.25 million (Figure 4). Estimation of Nef from microsatellite data using programs such as NeEstimator have not been reported, as these methods produced unreliable estimates and are known to be inappropriate for estimating the size of large populations, particularly with high levels of gene flow.

Bayesian skyline plots showing temporal changes in genetic diversity in Gray’s beaked whales (M. grayi) estimated from mitochondrial control region sequences. (a) Using the mutation rate from Dalebout et al. (2005). (b) Using the mutation rate from Ho et al. (2007, 2011). The x axis is in calendar years; the y axis is the product of effective population size and generation time (Nefτ). Grey shading indicates the 95% credibility intervals.
Discussion
We analysed samples collected from around the coast of New Zealand and Western Australia in the largest study on beaked whale population genetics to date. Our findings show that Gray’s beaked whales have high mitochondrial haplotype and nucleotide diversity relative to most beaked whales (Gray’s (530 bp): h=0.93–0.94, π=0.76–0.99%; Blainville’s (362 bp): h=0.87±0.07; π=0.49±0.35%; northern bottlenose (434 bp): h=0.57; π=0.15%), with the exception of the southern bottlenose whale (Hyperoodon planifrons) (238 bp): h=0.97; π=3.73% (Dalebout, 2002; Dalebout et al., 2001, 2005) (Table 3). Southern bottlenose whales also have a distribution extending throughout the Southern Ocean and have never been a target of whaling. The level of diversity observed in both these beaked whales contrasts with that found in pilot whales (Globicephala melas) and false killer whales (Pseudorca crassidens), where social factors are thought to contribute to low diversity (Whitehead, 1998). Spinner dolphins (Stenella longirostris longirostris) in the waters of French Polynesia are also pelagic, with a distribution concentrated around particular island groups and significant gene flow between these areas (Oremus et al., 2007). Our diversity statistics are comparable to this species and it is likely that Gray’s beaked whales show similar levels of movement and gene flow.
Our estimation of Fu’s Fs is negative and highly significant, indicating a population expansion or a selective sweep. A rapid radiation of Gray’s beaked whales during their divergence from the most recent common ancestor could potentially explain these signatures. However, the phylogeny of the ziphiids is currently in question as new and more informative genomic markers enable its revision (Morin et al., 2013). Gray’s beaked whales are unlikely to have undergone any recent genetic population bottleneck, although our data reflect long-term historical demographic patterns, and cannot determine more recent population changes. This species has no documented history of human consumption in this region and, therefore, these results are perhaps unsurprising (Robards and Reeves, 2011). However, there is the potential that a pelagic species such as Gray’s beaked whales is impacted upon by fisheries by-catch and, given the difficulties in carcass recovery and species identification, assessment data is currently unavailable (Madsen et al., 2014). To detect more recent population changes both census data and alternative genetic markers would be required, and current by-catch rates would be helpful in assessing potential human-induced mortality.
Our study is limited by small sample size from Australia, and therefore our results comparing Gray’s beaked whale population structure across to New Zealand are preliminary. However, in general, our analyses of data from both mtDNA and microsatellite markers indicate a lack of genetic structure across the ~6000km-wide study area. None of the pairwise comparisons of genetic differentiation based on Fst were significant at the P<0.05 level, and therefore our results are consistent with a single Gray’s beaked whale population. However, further samples from Australia are needed to confirm these findings.
Overall, this result contrasts with our original hypothesis predicting restricted gene flow between east and west coasts of New Zealand. Studies of population structure in beaked whales are inherently difficult because of the paucity of material available for genetic analysis; however, in northern bottlenose whales significant genetic structure was detected across a distance of ~2000 km between The Gully, off Nova Scotia, and the Labrador Sea (Dalebout et al., 2006). This structure is thought to result from a combination of habitat specificity, that is, the need to associate with submarine canyons, and a genetic bottleneck because of hunting (Dalebout et al., 2001). Cuvier’s and Blainville’s beaked whales are both cosmopolitan species that are broadly distributed throughout the world’s oceans. These species show clear differentiation between ocean basins, with little contemporary interoceanic gene flow (Dalebout et al., 2005; Morin et al., 2013). This differentiation is thought to reflect patterns of long-term divergence as a result of the species’ radiation, habitat preferences and/or social organization. Such genetic structure is not unusual for marine organisms with either site fidelity to breeding grounds, for example, white sharks (Carcharodon carcharias) (Bonfil et al., 2005), or feeding grounds, for example, herring (Clupea harengus) (Gaggiotti et al., 2009).
In contrast, given the results of our study, interoceanic gene flow is highly likely in the case of Gray’s beaked whales, particularly as there are no large continents that restrict movement throughout their distribution. This pattern is the first described in the genus Mesoplodon, and while our samples cover approximately one-third of the species’ range, further samples are needed from South Africa, South America and the Southern Ocean to confirm this finding. There are both fish and squid species that exhibit similar levels of connectivity across comparable spatial scales (e.g., orange roughy (Hoplostethus atlanticus), Varela et al., 2012; giant squid (Architeuthis spp.), Winkelmann et al., 2013) and it is likely that there are aspects of these species’ population biology that are common.
Thompson et al. (2013) suggests that, given stranding patterns, seasonal shifts in distribution associated with the calving season are likely in Gray’s beaked whales, perhaps in relation to a dependency on inshore waters. However, should these preferences exist they are clearly not driving long-term genetic differentiation or there is enough habitat of sufficient quality within the study area to support multiple calving grounds. Interestingly, the morphological differences seen in Gray’s beaked whales between the east and west coasts of New Zealand are not reflected in the genetic data (Thompson et al., 2014). This suggests that such morphological differences occur in the presence of gene flow and could perhaps result from phenotypic plasticity and/or dietary preferences. There are several examples of such phenotypic plasticity in cetaceans, for example, bottlenose dolphins (Tursiops truncatus) (Viaud-Martinez et al., 2008) and killer whales (Foote et al., 2009). These examples are thought to indicate ecological differences and are accompanied by an associated genetic divergence, but this is not the case in our study.
Given the degree of genetic homogeneity found between all regions, these results suggest that it is unlikely that the whales found off the coast of Western Australia are distinct from those found around the coast of New Zealand. We speculate that Gray’s beaked whales may move freely between these areas perhaps following the subtropical convergence, the boundary between cold sub-Antarctic and warmer subtropical waters, that dominates the centre of this species distribution (Garner, 1959; Heath, 1981). A number of marine mammal species are known to take advantage of this convergence, which is associated with areas of high primary productivity. Sightings surveys off the coast of south Australia have detected several beaked whales (Gill et al., 2015) with one particular sighting involving a single group of 20 unidentified mesoplodonts largely fitting the description of Gray’s beaked whales (P. Gill, pers. comm.). It is highly possible that such an oceanographic feature, which can be as productive as the Benguela Upwelling (van Ruth et al., 2010), may facilitate movements of Gray’s beaked whales and act as a gene flow ‘conveyer belt’ between New Zealand and Australia.
The panmictic pattern in Gray’s beaked whales may also be a result of social factors that promote gene flow, as has been suggested in common dolphins in the North Atlantic. Gray’s are unique among the beaked whales in that they commonly strand in groups. The holotype specimen was one of 28 animals stranded in the Chatham Islands in 1875 (von Haast, 1876), and other large strandings (4–6 animals) occur frequently around New Zealand (New Zealand Department of Conservation, unpublished data). It has been proposed that these larger strandings are breeding aggregations as in some cases they include multiple adult males, although further behavioural evidence would be required to confirm this (Dalebout, 2002). Whether these larger groups are formed for the purpose of mating is unknown but it is possible that Gray’s beaked whales have a mating system that is distinct from other ziphiids and more akin to what has been described in the delphinids. The high levels of genetic diversity and a lack of differentiation across the geographical range of our study may imply a promiscuous and/or polygynous mating system that promotes gene flow.
There are many limitations to estimates of female effective population size; several assumptions of the coalescent model are violated because of the lack of basic knowledge of this species biology. However, based on mitochondrial data, our analyses imply that Gray’s beaked whales have existed as an increasing population with no historical population bottleneck.
Given a plausible generation time for Gray’s beaked whales of 15 years, as is estimated for Cuvier’s beaked whales (Dalebout et al., 2005) and spinner dolphins (Oremus et al., 2007), our estimate of mean female effective population size ranges from 676 000 (26 000–3.45 million, 95% CI) to a lower estimate of 30 600 (1333–150 000, 95% CI). In all estimates, our credibility intervals highlight the high degree of uncertainty, and upper limits of female effective population size of whales do not generally reach into the millions, for example, Cuvier’s beaked whales in the Southern Ocean have an upper limit of 189 000 (Dalebout et al., 2005) and the minke whale (Balaenoptera acutorostrata) upper limit is 800 000 (Alter and Palumbi, 2009). Although we have applied a coalescent approach, which tends to be more accurate than deterministic methods, there are still considerable limitations. Effective population size estimates are strongly influenced by the mutation rate, with underestimation of rates resulting in large overestimates in population size (Luikart et al., 2010). In general, Nef is most difficult to estimate in large populations with moderate gene flow and this difficulty can lead to extremely large confidence intervals as are seen in our estimates (Hare et al., 2011). In this context, we suggest that our estimates of Nef should be considered as indicative of a large population with no bottleneck. This contradicts the basic assumption that, in general, beaked whales exist at naturally low abundances and, hence, are rarely observed at sea (Pitman, 2009). In the case of Gray's beaked whales, the rarity in sightings is more likely due to their offshore distribution and cryptic behaviour, together with a paucity of dedicated oceanic surveys.
In conclusion, our results suggest that Gray’s beaked whales form a large panmictic population. It is most likely that significant genetic connectivity exists between the waters of New Zealand and Western Australia. Although there are limitations in our sampling, and consequently our analyses, our inability to detect genetic heterogeneity throughout the study area suggests that there is an absence of any particular habitat dependencies, social factors or historical population depletion that have restricted gene flow. We suggest, given the strength of our findings, that Gray’s beaked whales in New Zealand and Australian waters be managed as a single management unit. Our study highlights the value of long-term stranding collections in studying populations of elusive, long-lived, slow-breeding species. With more extensive sampling, and higher resolution genetic markers (e.g., single-nucleotide polymorphisms), we suggest that future research that helps to elucidate any cryptic population structure in Gray’s beaked whales would be a valuable contribution to the study of this species.
Data archiving
Reference DNA sequences are available under GenBank accession nos. KJ767593–KJ767630. The genotype–haplotype assignments and information about sample location are available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.f47f6.
References
Aasen E, Medrano JF . (1990). Amplification of the ZFX and ZFY genes for sex identification in humans, cattle, sheep and goats. Nat Biotech 8: 1279–1281.
Alter SE, Palumbi SR . (2009). Comparing evolutionary patterns and variability in the mitochondrial control region and cytochrome b in three species of baleen whales. J Mol Evol 68: 97–111.
Alter SE, Ramirez SF, Nigenda S, Ramirez JU, Bracho LR, Palumbi SR . (2009). Mitochondrial and nuclear genetic variation across calving lagoons in eastern North Pacific gray whales (Eschrichtius robustus. J Hered 100: 34–46.
Ayers KL, Waters JM . (2005). Marine biogeographic disjunction in central New Zealand. Mar Biol 147: 1045–1052.
Baker CS, Slade RW, Bannister RB, Abernethy RB, Weinrich MT, Lien J et al. (1994). Hierarchical structure of mitochondrial DNA gene flow among humpback whales, Megaptera novaeangliae, world-wide. Mol Ecol 3: 313–327.
Bilgmann K, Möller LM, Harcourt RG, Gales R, Beheregaray LB . (2008). Common dolphins subject to fisheries impacts in Southern Australia are genetically differentiated: implications for conservation. Anim Conserv 11: 518–528.
Bonfil R, Meȳer M, Scholl MC, Johnson R, O’Brien S, Oosthuizen H et al. (2005). Transoceanic migration, spatial dynamics, and population linkages of white sharks. Science 310: 100–103.
Boschema H . (1950). Maxillary teeth in specimens of Hyperoodon rostratus (Muller) and Mesoplodon grayi Von Haast stranded on the Dutch coasts. Koninklijke Nederlandse Akademie van Wetenschappen 53: 775–786.
Butler A, Althaus F, Furlani D, Ridgway K . (2002) Assessment of the conservation values of the Bonney Upwelling. A component of the Commonwealth Marine Conservation Assessment Program 2002–2004. CSIRO report to Environment Australia. Hobart, Tasmania, Australia: CSIRO Marine and Atmospheric Research.
Chivers SJ, Baird RW, McSweeney DJ, Webster DL, Hedrick NM, Salinas JC . (2007). Genetic variation and evidence for population structure in eastern North Pacific false killer whales (Pseudorca crassidens. Can J Zool 85: 783–794.
Constantine R, Carroll E, Stewart R, Neale D, van Helden A . (2014). First record of True’s beaked whale Mesoplodon mirus in New Zealand. Mar Biodivers Rec 7: e1.
Dalebout ML . (2002). Species identity, genetic diversity and molecular systematic relationships among the Ziphiidae (beaked whales). PhD thesis, University of Auckland, New Zealand.
Dalebout ML, Hooker SK, Christensen I . (2001). Genetic diversity and population structure among northern bottlenose whales, Hyperoodon ampullatus, in the western North Atlantic Ocean. Can J Zool 79: 478–484.
Dalebout ML, Robertson KM, Frantzis A, Engelhaupt D, Mignucci-Giannoni AA, Rosario-Delestre RJ et al. (2005). Worldwide structure of mtDNA diversity among Cuvier's beaked whales (Ziphius cavirostris: implications for threatened populations. Mol Ecol 14: 3353–3371.
Dalebout ML, Russell KG, Little MJ, Ensor P . (2004). Observations of live Gray's beaked whales (Mesoplodon grayi in Mahurangi Harbour, North Island, New Zealand, with a summary of at-sea sightings. J R Soc NZ 34: 347–356.
Dalebout ML, Ruzzante DE, Whitehead H, Øien NI . (2006). Nuclear and mitochondrial markers reveal distinctiveness of a small population of bottlenose whales (Hyperoodon ampullatus in the western North Atlantic. Mol Ecol 15: 3115–3129.
De Leo FC, Smith CR, Rowden AA, Bowden DA, Clark MR . (2010). Submarine canyons: hotspots of benthic biomass and productivity in the deep sea. Proc R Soc Ser B 277: 2783–2792.
Drummond AJ, Suchard MA, Xie D, Rambaut A . (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29: 1969–1973.
Earl DA, vonHoldt BM . (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Res 4: 359–361.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Lischer HEL . (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10: 564–567.
Foote AD, Newton J, Piertney SB, Willerslev E, Gilbert MTP . (2009). Ecological, morphological and genetic divergence of sympatric North Atlantic whale populations. Mol Ecol 18: 5207–5217.
Fu XY . (1997). Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147: 915–925.
Gaggiotti OE, Bekkevold D, Jørgensen HBH, Foll M, Carvalho GR, Andre C et al. (2009). Disentangling the effects of evolutionary, demographic, and environmental factors influencing genetic structure of natural populations: Atlantic herring as a case study. Evolution 63: 2939–2951.
Garner DM . (1959). The sub-tropical convergence in New Zealand surface waters. New Zeal J Geol Geop 2: 315–337.
Gill PC, Pirzl R, Morrice MG, Lawton K . (2015). Cetacean diversity of the continental shelf and slope off Southern Australia. J Wildl Manage 79: 672–681.
Gilson A, Syvanen M, Levine K, Banks J . (1998). Deer gender determination by polymerase chain reaction: validation study and application to tissues, bloodstains, and hair forensic samples from California. Calif Fish Game 84: 159–169.
Graves JA, Helyar A, Biuw M, Jüssi M, Jüssi I, Karlsson O . (2008). Microsatellite and mtDNA analysis of the population structure of grey seals (Halichoerus grypus from three breeding areas in the Baltic Sea. Conserv Genet 10: 59–68.
Hare MP, Nunney L, Schwartz MK, Ruzzante DE, Burford M, Waples RS et al. (2011). Understanding and estimating effective population size for practical application in marine species management. Conserv Biol 25: 438–449.
Heath RA . (1981). Oceanic fronts around Southern New Zealand. Deep-Sea Res 28: 547–560.
Ho SYW, Kolokotronis S-Y, Allaby RG . (2007). Elevated substitution rates estimated from ancient DNA sequences. Biol Lett 3: 702–705.
Ho SYW, Lanfear R, Phillips MJ, Barnes I, Thomas JA, Kolokotronis S-O et al. (2011). Bayesian estimation of substitution rates from ancient DNA sequences with low information content. Syst Biol 60: 1–10.
Hoelzel AR, Dahlheim M, Stern SJ . (1998). Low genetic variation among killer whales (Orcinus orca in the eastern North Pacific and genetic differentiation between foraging specialists. J Hered 89: 121–128.
Hudson RR . (1990) Gene genealogies and the coalescent process. In: Futuyama F, Antonovics JD (eds). Oxford Surveys in Evolutionary Biology, Vol 7. Oxford University Press: New York, NY, USA.
Huelsenbeck JP, Ronquist F . (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17: 754–755.
Jackson JA, Baker CS, Vant M, Steel DJ, Medrano-González L, Palumbi SR . (2009). Big and slow: Phylogenetic estimates of molecular evolution in baleen whales (suborder Mysticeti. Mol Biol Evol 26: 2427–2440.
Jakobsson M, Rosenberg NA . (2007). CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23: 1801–1806.
Johnson M, Hickmott LS, Aguilar Soto N, Madsen PT . (2008). Echolocation behaviour adapted to prey in foraging Blainville’s beaked whale (Mesoplodon densirostris. Proc R Soc Ser B 275: 133–139.
Jombart T . (2008). Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24: 1403–1405.
Katoh K, Misawa K, Kuma K, Miyata T . (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30: 3059–3066.
Kalinowski ST . (2005). HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5: 187–189.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28: 1647–1649.
Luikart G, Ryman N, Tallmon DA, Schwartz MK, Allendorf FW . (2010). Estimation of census and effective population sizes: the increasing usefulness of DNA-based approaches. Conserv Genet 11: 355–373.
MacLeod CD, Santos MB, Pierce GJ . (2003). Review of data on diets of beaked whales: evidence of niche separation and geographic seqregation. J Mar Biol Assoc UK 83: 651–665.
MacDiarmid AB, Law CS, Pinkerton M, Zeldis J . (2013) New Zealand marine ecosystem services. In: Dymond JR (ed). Ecosystem Services in New Zealand—Conditions and Trends. Manaaki Whenua Press: Lincoln, New Zealand.
Madsen PT, Aguilar de Soto N, Tyack PL, Johnson M . (2014). Beaked whales. Curr Biol 24: R728–R730.
McSweeney D, Baird R, Mahaffy S . (2007). Site fidelity, associations, and movements of Cuvier's (Ziphius cavirostris and Blainville's (Mesoplodon densirostris beaked whales off the island of Hawai‘i. Mar Mammal Sci 23: 666–687.
Meirmans PG, Hedrick PW . (2011). Assessing population structure: F st and related measures. Mol Ecol Resour 11: 5–8.
Meirmans PG, Van Tienderen PH . (2004). GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4: 792–794.
Moore JE, Barlow JP . (2013). Declining abundance of beaked whales (Family Ziphiidae) in the California current large marine ecosystem. PLoS One 8: e52770.
Morin PA, Archer FI, Foote AD et al. (2010). Complete mitochondrial genome phylogeographic analysis of killer whales (Orcinus orca indicates multiple species. Genome Res 20: 908–916.
Morin PA, Duchene S, Lee N, Durban J, Claridge D . (2013). Preliminary analysis of mitochondrial genome phylogeography of Blainvilles’, Cuvier’s and Gervais’ beaked whales. Report to the Scientific Committee of the International Whaling Commission (No. SC/64/SM14) p 17.
New LF, Moretti DJ, Hooker SK, Costa DP, Simmons SE . (2013). Using energetic models to investigate survivial and reproduction of beaked whales (family Ziphiidae). PLoS ONE 8: e68725.
Oremus M, Gales R, Dalebout ML, Funahashi N, Endo T, Kage T et al. (2009). Worldwide mitochondrial DNA diversity and phylogeography of pilot whales (Globicephala spp.). Biol J Linn Soc 98: 729–744.
Oremus M, Poole M, Steel D, Baker CS . (2007). Isolation and interchange among insular spinner dolphin communities in the South Pacific revealed by individual identification and genetic diversity. Mar Ecol Prog Ser 336: 257–289.
Patel S, Thompson K, Williams L, Tsai P, Constantine R, Millar C . (2014). Mining microsatellites for Gray’s beaked whale from second-generation sequencing data. Conserv Genet Resour 6: 657–659.
Pimper LE, Baker CS, Goodall RNP, Olavarría C, Remis MI . (2010). Mitochondrial DNA variation and population structure of Commerson’s dolphins (Cephalorhynchus commersonii in their southernmost distribution. Conserv Genet 11: 2157–2168.
Pitman R . (2009) Mesoplodont whales (Mesoplodon spp.). In: Perrin WF, Würsig B, Thewissen JGM (eds). Encyclopedia of Marine Mammals, 2nd edn. Academic Press: San Diego, CA, USA, pp 721–726.
Posada D . (2008). jModelTest: phylogenetic model averaging. Mol Biol Evol 25: 1253–1256.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Rambaut A, Suchard MA, Xie D, Drummond AJ . (2013). Tracer v1.5. Available from http://beast.bio.ed.ac.uk/Tracer accessed 12th May 2015.
Ramos-Onsins SE, Rozas J . (2002). Statistical properties of new neutrality tests against population growth. Mol Biol Evol 19: 2092–2100.
Robards MD, Reeves RR . (2011). The global extent and character of marine mammal consumption by humans: 1970–2009. Biol Conserv 144: 2770–2786.
Rosenberg NA . (2004). DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4: 137–138.
Sagnol O, Richter C, Field LH, Reitsma F . (2014). Spatio-temporal distribution of sperm whales (Physeter macrocephalus off Kaikoura, New Zealand, in relation to bathymetric features. NZ J Zool 41: 234–247.
Sambrook J, Fritsch EF, Maniatis T . (1989) Molecular Cloning: A Laboratory Manual, Vol 3. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA.
Scheidat M, Friedlaender A, Kock K-H, Lehnert L, Boebel O, Roberts J et al. (2011). Cetacean surveys in the Southern Ocean using icebreaker-supported helicopters. Polar Biol 34: 1513–1522.
Schick RS, Halpin PN, Read AJ, Urban DL, Best BD, Good CP et al. (2011). Community structure in pelagic marine mammals at large spatial scales. Mar Ecol Prog Ser 434: 165–181.
Schorr GS, Falcone EA, Moretti DJ, Andrews RD . (2014). First long-term behavioral records from Cuvier’s beaked whales (Ziphius cavirostris reveal record-breaking dives. PLoS One 9: e92633.
Sremba AL, Hancock-Hanser B, Branch TA, LeDuc RL, Baker CS . (2012). Circumpolar diversity and geographic differentiation of mtDNA in the critically endangered Antarctic blue whale (Balaenoptera musculus intermedia. PLoS One 7: e32579.
Tamura K, Nei M . (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10: 512–526.
Taylor BL, Baird R, Barlow J et al. (2008) Mesoplodon grayi. The IUCN Red List of Threatened Species 2008: e.T13247A3428839. Available at: www.iucnredlist.org (last accessed 27 February 2014).
Thompson K, Baker S, van Helden A, Patel S, Millar C, Constantine R . (2012). The world’s rarest whale. Curr Biol 22: 905–906.
Thompson K, Millar C, Baker C, Dalebout M, Steel D, van Helden A et al. (2013). A novel conservation approach provides insights into the management of rare cetaceans. Biol Conserv 157: 331–340.
Thompson K, Ruggiero K, Millar C, Constantine R, van Helden A . (2014). Large-scale multivariate analysis reveals sexual dimorphism and geographic differences in the Gray’s beaked whale. J Zool 294: 13–21.
Torres LG . (2013). Evidence for an unrecognised blue whale foraging ground in New Zealand. NZ J Mar Fresh 47: 235–248.
Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P . (2004). MICRO‐CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538.
Van Waerebeek K, Leaper R, Baker A, Papastavrou V, Thiele D, Findlay K et al. (2010). Odontocetes of the southern ocean sanctuary. J Cet Res Man 11: 315–346.
Varela AI, Ritchie PA, Smith PJ . (2012). Low levels of global genetic differentiation and population expansion in the deep-sea teleost Hoplostethus atlanticus revealed by mitochondrial DNA sequences. Mar Biol 159: 1049–1060.
van Ruth PD, Ganf GG, Ward TM . (2010). Hot-spots of primary productivity: an alternative interpretation to conventional upwelling models. Estuar Coast Shelf S 90: 142–158.
Viaud-Martinez KA, Brownell RL, Komnenou A, Bohonak AJ . (2008). Genetic isolation and morphological divergence of Black Sea bottlenose dolphins. Biol Cons 141: 1600–1611.
von Haast J . (1876). On a new ziphioid whale. Proc Zool Soc Lond 1876: 7–13.
Weilgart L . (2007). The impacts of anthropogenic ocean noise on cetaceans and implications for management. Can J Zool 85: 1091–1116.
Whitehead H . (1998). Cultural selection and genetic diversity in matrilineal whales. Science 282: 1708–1711.
Wimmer T, Whitehead H . (2004). Movements and distribution of northern bottlenose whales, Hyperoodon ampullatus, on the Scotian Slope and in adjacent waters. Can J Zool 82: 1782–1794.
Winkelmann I, Campos PF, Strugnell J, Cherel Y, Smith PJ, Kubodera T et al. (2013). Mitochondrial genome diversity and population structure of the giant squid Architeuthis: genetics sheds new light on one of the most enigmatic marine species. Proc R Soc Ser B 280: 20130273.
Wright AJ, Deak T, Parsons ECM . (2011). Size matters: management of stress responses and chronic stress in beaked whales and other marine mammals may require larger exclusion zones. Mar Pollut Bull 63: 5–11.
Acknowledgements
We thank the staff of the New Zealand Department of Conservation for tissue collection and database management, particularly L Boren and L Wakelin; local iwi and hapu, and Massey University necropsy teams. Samples from west Australia were supplied by: R O’Shea and J Bannister, Western Australian Museum; and N Gales, Australian Antarctic Division. We thank M Dalebout and D Steel for sample collection and archiving; A Stuckey, A Veale and R Bouckhaert for assistance with analyses; the Centre for Genomics, Proteomics and Metabolomics at the University of Auckland, and New Zealand Genomics Ltd. We also thank three anonymous reviewers for their useful comments. This project was funded by a University of Auckland Faculty Research Development Fund Grant 3702180 (CDM and RC). Samples are curated in the New Zealand Cetacean Tissue Archive at the University of Auckland under Department of Conservation Permit Rnw/HO/2009/03 and CITES institutional permit NZ010.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

Cite this article
Thompson, K., Patel, S., Baker, C. et al. Bucking the trend: genetic analysis reveals high diversity, large population size and low differentiation in a deep ocean cetacean. Heredity 116, 277–285 (2016). https://doi.org/10.1038/hdy.2015.99
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2015.99
This article is cited by
-
Evidence of historical isolation and genetic structuring among broadnose sevengill sharks (Notorynchus cepedianus) from the world’s major oceanic regions
Reviews in Fish Biology and Fisheries (2021)
-
The life aquatic: advances in marine vertebrate genomics
Nature Reviews Genetics (2016)
