
Abstract
The theory of island biogeography is most often studied in the context of oceanic islands where all island inhabitants are descendants from founding events involving migration from mainland source populations. Far fewer studies have considered predictions of island biogeography in the case of continental islands, where island formation typically splits continuous populations and thus vicariance also contributes to the diversity of island populations. We examined one such case on continental islands in southeastern Brazil, to determine how classic island biogeography predictions and past vicariance explain the population genetic diversity of Thoropa taophora, a frog endemic to the Atlantic Coastal Forest. We used nuclear microsatellite markers to examine the genetic diversity of coastal and island populations of this species. We found that island isolation has a role in shaping the genetic diversity of continental island species, with island populations being significantly less diverse than coastal populations. However, area of the island and distance from coast had no significant effect on genetic diversity. We also found no significant differences between migration among coastal populations and migration to and from islands. We discuss how vicariance and the effects of continued migration between coastal and island populations interact to shape evolutionary patterns on continental islands.
Similar content being viewed by others


Introduction
Islands are excellent models for the study of evolutionary processes (MacArthur and Wilson, 1967; Losos and Schluter, 2000; Vellend, 2003). Numerous studies have focused on island systems to quantify the relative roles of colonization, extinction and isolation in shaping island diversity (Mayr, 1963; Jaenike, 1973; Abbott and Grant, 1976). These studies have typically examined the interplay of these processes as determinants of species richness (MacArthur and Wilson, 1967; Losos and Schluter, 2000; Whittaker et al., 2001; Ricklefs and Bermingham, 2004). However, more recently, island studies have focused on the roles of the parallel processes of drift and island isolation in the evolution and genetic differentiation of single species distributed on multiple islands (Frankham, 1997; Calsbeek and Smith, 2003; Vellend, 2003; Grazziotin et al., 2006; Jordan and Snell, 2008).
When populations of a species occur both on islands and on the coast, we can predict levels of intraspecific genetic diversity on islands based on their area and distance from coastal source populations (Jaenike, 1973; Frankham, 1997; Vellend, 2003). As in classic island biogeography theory, the predictions will vary with the dispersal ability of the organism. If dispersal to islands is rare, and hence island populations experience negligible migration, we expect lower genetic diversity in island populations relative to larger, coastal populations due to genetic drift (Frankham, 1997). Similarly, islands that are more distant from the coast should receive fewer migrants and less influx of genetic diversity. Therefore, the effects of drift will be intensified when founding populations are small, when islands are small in area and when islands are far from the mainland source population (Jaenike, 1973; Frankham, 1997; Clegg et al., 2002a; Velo-Antón et al., 2012).
Tests of these genetic predictions on island populations have been done primarily on oceanic islands (Frankham, 1997; Clegg et al., 2002a; Calsbeek and Smith, 2003). However, more recently studies have also focused on taxa inhabiting continental islands (Gill, 1980; Bittkau and Comes, 2005; Jordan and Snell, 2008; Velo-Antón et al., 2012). In contrast to oceanic islands that are formed de novo by volcanic activity, continental islands were once continuous with continental landmasses and are formed by sea level changes that isolate the highest points on the edge of the continental shelf. Therefore, the evolutionary dynamics of populations on continental islands are likely very different from those on oceanic islands. The formation of continental islands necessarily fragments species’ ranges; thus, although populations will be reduced in size and genetic drift will be increased due to population bottlenecks, the founding island population occurs through vicariance isolation, rather than a founding event by one or few immigrants from a mainland source population. Nonetheless, if the population remaining at the time of vicariance is small, and migration is sufficiently low, the predictions of island size and genetic diversity should hold, because smaller islands will support populations with lower sizes, and those populations will lose genetic diversity more rapidly due to drift (Frankham, 1996). Indeed, continental island populations examined to date show the expected pattern of reduced genetic diversity (Bittkau and Comes, 2005; Velo-Antón et al., 2012).
Migration patterns may also differ between continental and oceanic islands, because continental islands, by their nature, are generally closer to coastal landmasses. Migration between continental islands and the mainland may be more common, while species on oceanic islands that exist along an archipelago can exhibit gene flow among adjacent islands (Illera et al., 2007; Clegg and Phillimore, 2010; Illera et al., 2014; Bell et al., 2015a, 2015b). This distinction is a generalization; the migration levels in both oceanic and continental islands will depend on the dispersal ability of particular colonizing organisms and the degree to which marine environments act as barriers to dispersal. If marine dispersal is unlikely, then we predict that smaller islands and those further from the coast will receive fewer migrants, and as a result, genetic drift will be the dominant force shaping the genetic diversity of those isolated populations. Bittkau and Comes (2005) reported low rates of marine dispersal and high drift in the Aegean Nigella arvensis alliance, a group of annual plants distributed in the Aegean archipelago. However, if marine dispersal is sustained after isolation of continental islands, then we expect genetic diversity in island populations to approximate levels observed in populations on the coast.
Amphibians are poor dispersers across marine environments because of their inability to osmoregulate in saltwater (Duellman and Trueb, 1994). Therefore, the general assumption is that island populations of amphibians are completely isolated at the time of vicariance during formation of continental islands (Richards and Moore, 1996; Brown and Guttman, 2002). This isolation scenario seems to hold for some continental island dwelling amphibians: a recent study of fire salamanders showed a reduction of genetic diversity on insular populations indicating that these populations evolved in isolation without any subsequent marine dispersal events (Velo-Antón et al., 2012). However, some studies indicate that amphibians are in fact able to disperse to islands (Seppa and Laurila, 1999; Evans et al., 2003; Vences et al., 2003), although the exact mechanism of dispersal is unknown. In this study, we examine the population genetics of Thoropa taophora, a frog species endemic to the Atlantic Coastal Forest of Brazil (Feio et al., 2006) that inhabits rocky coastal shores and has some physiological tolerance to seawater (Bokermann, 1965; Sazima, 1971; Abe and Bicudo, 1991; Brasileiro et al., 2010).
We focused on a series of continental islands that flank the southeast coast of Brazil. These islands were formed by historical marine incursions due to glaciation events, tectonic activities and coastal erosion dynamics (Suguio and Martin, 1978). Geologic records show evidence of repeated incursions since the Pleistocene, with the last major incursion occurring in the past 15 000 years (Suguio et al., 2005). During this last transgressive phase, coastal sea levels rose over 100 meters to reach present levels (Suguio et al., 2005). This incursion isolated present-day continental islands and likely fragmented the ranges of several Atlantic Coastal Forest species, including our focal species. Studies of other species endemic to this region show a reduction of genetic diversity in island species or populations relative to those on the mainland (Grazziotin et al., 2006; Bell et al., 2012). Here, we build on these studies by investigating fine-scale genetic diversity in an organism that exhibits tolerance to saltwater and thus may be capable of dispersing to continental islands.
We used nuclear microsatellite markers to examine the genetic diversity of coastal and island populations of Thoropa taophora. Our goals were to quantify (1) differences in genetic diversity of populations along the coast and on continental islands due to past vicariance and island isolation, (2) the genetic consequences of island area and distance from coast and (3) marine migration rates between coastal and island populations. Our study focuses on the microevolutionary processes contributing to the genetic patterns in continental island populations. We discuss our results in light of processes that are specific to continental islands, including the extent of genetic bottlenecks during population vicariance and the potential genetic contributions resulting from migration from nearby coastal populations.
Materials and Methods
Study species and population sampling
Thoropa taophora is a frog endemic to the Atlantic Coastal Forest of Brazil, where it occurs from sea level to ~1000 m in elevation (Feio et al., 2006). Populations of T. taophora above 700 m are very rare (C.Brasileiro—personal obs.) or have disappeared in recent times (Heyer et al., 1990), thus most populations are found at or near sea level. Past work on this genus indicates that these frogs have unique natural histories: they live and breed on rocky marine shores (Giaretta and Facure, 2004), tadpoles are semiterrestrial and develop in seeps and streams (Bokermann, 1965; Rocha et al., 2002), they have high tolerance to short exposures to salt water (Abe and Bicudo, 1991) and they eat marine invertebrates and have physiological adaptations for excreting salt (Bokermann, 1965; Sazima, 1971; Brasileiro et al., 2010).
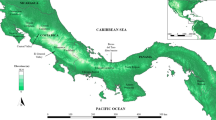
We sampled Thoropa taophora at 17 localities (Figure 1), including 7 coastal sites and 10 sites on continental islands. Our sampling covered most of the range of T. taophora. The choice of islands was based on the presence of suitable habitat (forest and the presence of freshwater seeps on rocky shores) and by our ability to safely access them. Each locality was visited at least three times between April 2004 and April 2007 for the collection of a random sample of adults and tadpoles. At each locality, we collected at a single stream or coastal rock outcrop. To avoid potential collection of siblings, we collected a maximum of two tadpoles per stream and each tadpole was collected from a different reach of the stream. Tissues were collected in the form of toe clips or liver samples from adults or whole tadpoles in the case of larval samples. All samples were preserved in 95% ethanol for later use in genetic analyses. Voucher specimens have been deposited in the Coleção de Anuros, Universidade Estadual Paulista Rio Claro, Brazil.

Localities for Thoropa taophora population samples in our study. Maps A and B show relative locations of populations in the southeastern and northeastern coastal regions of São Paulo state and the genetic composition of each population. Asterisks (*) indicate island localities. Bayesian assignment placed individuals into eight genetic demes, each represented by a unique color. Each bar indicates an individual’s proportional membership to those demes.
Laboratory protocols
Whole-genomic DNA was extracted from tissues in 150 μl of a 5% Chelex-100 solution (Bio-Rad, Hercules, CA, USA) with 1 μl Proteinase K (20 mg ml−1) by incubating at 55 °C for 180 min and denaturing at 99° C for 10 min. Chelex supernatants were used directly as a template in PCR reactions. We used nine polymorphic microsatellite loci previously characterized for this species (Duryea et al., 2008). We amplified all loci in a total volume of 10 μl with 1 μl template DNA, 1 × Taq buffer (Roche, Indianapolis, IN, USA), 1.5 mM MgCl2, 0.4 mM dNTPs, 0.2 μM of each primer (forward and reverse) and 0.25 U of Taq polymerase (Roche) under amplification conditions optimized for each locus (Duryea et al., 2008). After amplification, we pooled PCR products for each individual into three groups (Duryea et al., 2008) and combined 1 μl of pooled PCR product with 18.85 μl Hi-Di formamide (Applied Biosystems, Foster, CA, USA) and 0.15 μl GeneScan-500 LIZ for genotyping on a 3100 Genetic Analyzer (Applied Biosystems). Genotypes were assigned to pre-determined size class bins using GENEMAPPER 3.5 (Applied Biosystems).
Genetic diversity
We tested for Hardy–Weinberg equilibrium (HWE) and linkage disequilibrium (LD) at each locus across all populations using FSTAT 2.9.3 (Goudet, 1995). Deviations from HWE were tested within populations based on 1000 randomizations, and the FIS statistic was used to assess significant differences between randomized and observed data at a table-wide α level of 0.05 (adjusted P-value=0.00031). LD was tested based on 10 000 randomizations and pairs of loci were determined to be in disequilibrium at an adjusted P-value of 0.000082. We tested for the presence of null alleles using MICRO-CHECKER 2.2.3 (Van Oosterhout et al., 2004). A large proportion of data (>50% of sampled individuals) were missing for a single locus (TmT27) in four of our sampled populations (Prumirim, Tamanduá, Couves Sul and Juréia); thus, we excluded this locus from further distance-based analyses so as not to bias our results. However, we retained these data for assignment tests and Bayesian estimates of migration, which are more robust to the presence of missing data (Carlsson, 2008).
We quantified genetic diversity at each population using various indices: mean number of alleles estimated in FSTAT 2.9.3 (Goudet, 1995), mean allelic richness estimated in HP-RARE (Kalinowski, 2005), and observed and expected levels of heterozygosity in ARLEQUIN 3.1.1 (Excoffier et al., 2005). HP-RARE uses rarefaction and a hierarchical sampling design to adjust allelic richness for unequal sample sizes among sampling sites.
We performed statistical tests in R 3.1.0 (R Core Team, 2014) to compare levels of genetic diversity among coastal and island populations. We compared the average observed heterozygosity and average allelic richness between coastal and island populations using one-way ANOVA. For both observed herterozygosity and allelic richness, we used Bartlett’s test to test for homogeneity of variance between coastal and island measures of diversity. We also conducted four linear regressions in R 3.1.0 (R Core Team, 2014) to test for the effects of island area and distance from coast on observed heterozygosity and average allelic richness. Area, distance and diversity estimates were log10 transformed for normalization. As Ilhabela is more than two orders of magnitude larger than the others islands, we did all analyses with and without the samples from this island to control for potential bias based on its inclusion.
Population differentiation
To test for genetic differentiation among populations, we calculated pairwise FST using ARLEQUIN 3.1.1 (Excoffier et al., 2005). Tests for significant differences between populations were based on 1000 permutations of haplotypes and α=0.05. We also calculated F′ST, a standardized measure of population differentiation that uses the AMOVA framework and accounts for variation among sample sizes, using 999 bootstraps in the program GenoDive 2.0b27 (Meirmans and Van Tienderen, 2004).
We compared FST values among island and coastal populations to determine the significance of marine barriers in the genetic differentiation of T. taophora. We grouped FST values in three different categories: those estimated between two coastal populations, between one coastal and one island population, and between two island populations. These categories were defined a priori based on the geographic location of our localities (that is, whether they occurred on an island or continental landmass). A one-way ANOVA was conducted in the program JMP 7.1 (SAS Institute) to test for significant differences among mean FST values, and multiple comparisons using the Tukey-Kramer Honestly Significant Difference method were used to determine which categories of FST values differed.
As an additional comparison of population differentiation, we conducted a hierarchical AMOVA in the program ARLEQUIN 3.1.1 (Excoffier et al., 2005), in which we compared variation between two groups (populations grouped as island or coastal), among populations within groups, and within populations. This hierarchical framework allowed us to determine how variation is distributed across our sampled populations and to what degree the different levels proportionally describe the genetic diversity of sampled individuals.
We used the program STRUCTURE 2.0 (Pritchard et al., 2000) to infer the number of genetic demes in our sampled populations and the degree of admixture among them. We implemented an admixture model with correlated allele frequencies among populations. We performed 20 runs at each level of genetic clustering (K ranging from 1 to 20), with 3 million repetitions following a burn-in of 1 million steps. Log-likelihood scores for this range of K values were plotted following the method of Evanno et al. (2005). The distribution of log-likelihood scores showed a pattern of incremental increase with successively larger values of K, with an asymptote in the K range of 6 to 10. We created graphical outputs at each K for the run with the highest LnP(D) using the program DISTRUCT (Rosenberg, 2004). We examined these plots to determine the level of K, in the range from 6 to 10, at which adding new clusters resulted in heterogeneity within single geographic locality samples, rather than genetic demes corresponding to specific localities or geographic regions. This value of K is the most parsimonious estimate, given the geographic arrangement of populations, and was taken as the best representation of genetic structure for the populations in our study.
Island population sizes, migration rates and divergence times
We used the isolation with migration model implemented in the program IM (Hey and Nielsen, 2004) to estimate relative population sizes, levels of migration and divergence times for selected pairs of populations. Demographic parameters of island populations were compared with the nearest sampled coastal populations, which were selected a priori as the ancestral population. We also compared three pairs of coastal populations in northern, central and southern sections of our sampling range as a metric of migration rates and divergence times expected among contiguous populations in the Atlantic Forest. The two populations sampled on Ilhabela were combined for IM analyses, as we determined them to belong to the same genetic deme.
IM applies a Bayesian likelihood analysis to fit an isolation-with-migration model to a pair of populations to estimate historical demographic parameters for the two populations. A Markov chain Monte Carlo approach with the SMM mutation model was used to obtain maximum likelihood estimates of model parameters. Seven of the nine loci were selected for IM analysis; loci TmD48 and TmD49 were excluded because they contained complex repeat motifs and did not follow the stepwise mutation model (Duryea et al., 2008). The model incorporated six demographic parameters scaled by mutation rate (μ): ancestral effective population size (θa=2Neμ), effective population sizes for the two descendent populations (θ1 and θ2), time since population divergence (t=tμ, where t is measured in generations), and directional migration rates (m1 and m2=m/μ), where m1 represents migration into population 1 from population 2, as you move forward in time, and m2 represents mutation scaled migration from population 1 into population 2. We used a geometric heating scheme for all of our comparisons, but priors varied among our tests (for estimates that reached converge, priors ranged as follows: θa=2–4, θ1=0.03–0.7, θ2=0.03–0.5, t=0.05–5, m1=1–30 and m2=3–75). We used preliminary simulations to determine the appropriate priors and run conditions for the Markov chain Monte Carlo. In each case, we performed a final run of 60 million generations or more to ensure convergence of parameter estimates. Individual simulations were run at least three times with a different random seed to ensure similar distributions for parameter estimates.
Of the six parameters estimated in IM, we focused on five to infer demographic patterns between island and coastal populations: effective population sizes of islands since divergence from coastal populations (θI), effective ancestral population size from which island populations split (θA), divergence times of islands from coastal populations (t) and migration rates into coastal and island populations (m1 and m2). Population migration rates (2Nm) were calculated from IM parameters using the formula: 2Nm=θm/2 (Hey and Nielsen, 2004). The mutation rate for microsatellite loci and exact generation time for this frog are not known; therefore, we used an average microsatellite mutation rate of 10−5 and an average generation time for frogs of 4 years (Ellegren, 2000; Rowe and Beebee, 2007) to calculate estimates of effective population size (N) and time since splitting (t) from IM parameters of θ and t, respectively. Thus, our estimates of population migration rates, effective population sizes and times since divergence are not to be taken as exact values but rather to be used for comparative purposes among populations.
We used JMP 7.1 (SAS Institute, 1995) to perform four regressions to test for relationships between (1) island effective population sizes and island area, (2) island effective population sizes and distance from coast, (3) migration rates to islands and island area, and (4) migration rate to islands and distance from coast. Area, distance, population migration rates and effective population size were log10 transformed for normalization. The populations from Ilhabela were excluded from the analyses of area because that island is more than two orders of magnitude larger than the others.
Results
Population sampling
On average we collected 25.8 (range 9–49) individuals at each locality (Table 1). We sampled islands ranging in area from 6 to 27 000 ha (mean=3038±8986 ha; Table 2). Excluding Ilhabela, islands ranged in area from 6 to 110 ha (mean=43±34 ha, Table 2). The smallest distance to the coast for these islands ranged from 0.50 to 2.46 km (mean=1.43 km). For island populations used in IM analysis, the distance to the nearest sampled coastal population ranged from 3.7 to 39.7 km (mean=14.3 km; Table 2).
Genetic diversity
FSTAT randomization tests revealed no pairs of loci in LD across all populations, although a few were in disequilibrium within a few populations. The FIS analyses also showed none of the loci to have excess heterozygosity, although six populations showed heterozygotes deficiency (Table 1). None of the populations deviated from HWE across all loci. Because we detected no loci that consistently deviated from HWE or showed linkage disequilibrium, we retained all loci for downstream analyses.
MICRO- CHECKER 2.2.3 (Van Oosterhout et al., 2004) detected an excess of homozygosity in six of the nine microsatellite loci (TmD48, TmT27, TmD8, TmD49, TmD31 and TmD21). However, no locus showed a consistent excess of homozygosity across all populations. Although an excess of homozygosity may indicate the presence of null alleles, we retained all loci for analyses because it is impossible to distinguish the presence of null alleles from true excess of homozygosity (that can occur due to population structure and drift) without additional genetic information for this species. Given that our sampling included island populations that have certainly evolved under limited gene flow with the mainland, we felt that eliminating markers would cause unnecessary loss of power for our analyses.
Our analyses of intrapopulation genetic diversity revealed consistent differences between island and coastal populations. Bartlett’s test confirmed homogeneity of variance for observed heterozygosity (K21=0.552, P=0.46) and allelic richness (K21=0.152, P=0.70) between coastal and island estimates, thus meeting the assumptions for ANOVA. We found significantly higher genetic diversity in coastal populations than island populations. Coastal populations have significantly higher mean observed heterozygosity (ANOVA; F1,17=13.34; P<0.0024; Figure 2) and mean allelic richness (ANOVA; F1,17=40.79; P<0.0001; Figure 2). However, the coastal population of Juréia and the island population of Ilhabela were exceptions; Juréia showed lower levels of diversity than most coastal populations and Ilhabela showed higher levels than most island populations (Table 1).

Means and s.e. values for (a) observed heterozygosity and (b) allelic richness among coastal and island populations of Thoropa taophora. Coastal populations have significantly higher mean observed heterozygosity and mean allelic richness.
Despite these differences among coastal and island populations, genetic diversity of island populations did not vary with island area or distance from the mainland. Linear regressions (excluding Ilhabela) showed no effect of island area on mean observed heterozygosity (ANOVA; F1,7=0.2978; P=0.605) or allelic richness (ANOVA; F1,7=0.4335; P=0.5347). Similarly, island distance from the coast had no effect on mean observed heterozygosity (ANOVA; F1,7=0.7873; P=0.4091) or mean allelic richness (ANOVA; F1,7=2.198; P=0.1887). Linear regressions including Ilhabela were consistent: we find no effect of island area on mean observed heterozygosity (ANOVA; F1,8=0.7342; P=0.4199) or allelic richness (ANOVA; F1,8=3.42; P=0.1069). Similarly, island distance from the coast had no effect on mean observed heterozygosity (ANOVA; F1,8=0.4407; P=0.5281) or mean allelic richness (ANOVA; F1,8=3.446; P=0.1058).
Population differentiation
FST values were high among all T. taophora populations, and all pairwise comparisons of populations were significantly different than zero (Supplementary Table S1). Bartlett’s test confirmed homogeneity of variance for the three classes (C/C, C/I, and I/I) of FST values (K22=0.4002, P=0.82). All three categories of FST comparisons (coastal/coastal, coastal/island, and island/island) were significantly different from one another (ANOVA; F2,135=53.76; P<0.0001; Figure 3). Coastal populations showed the lowest level of differentiation (mean FST=0.1908±0.1104). Comparing only island populations showed the highest divergences (mean FST=0.4410±0.09985), and coastal/island comparisons showed intermediate but also high differentiation (mean FST=0.3009±0.09218) (Figure 3). The single exception to this pattern was the coastal population of Juréia that was highly differentiated from all populations, including other coastal ones (Supplementary Table S1).

Means and s.e. values for population divergence (FST values) between coastal populations (C/C), coastal and island populations (C/I), and island populations (I/I). The three groups of FST estimates were significantly different by a Tukey–Kramer HSD test (significant differences indicated by letters [a], [b] and [c]), with C/C populations showing the lowest divergence and I/I populations showing the highest.
F′ST values (standardized measure of population differentiation that uses the AMOVA framework) showed similar trends to FST estimates (Supplementary Table S2). Coastal populations showed the lowest level of differentiation (mean F′ST=0.5253±0.2256), island populations showed the highest divergences (mean F′ST=0.7575±0.1238) and coastal/island comparisons showed intermediate differentiation (mean F′ST=0.6816±0.1590) (Supplementary Table S2).
The hierarchal AMOVA analysis showed significant variation among (AMOVA; P<0.001) and within populations (AMOVA; P<0.001), but not among groups of island and coastal populations (AMOVA; P=0.067). The greatest genetic variation is explained within populations (54%, FST=0.4634), 34% of variation was explained among populations (FSC=0.3890) and 12% of variation was explained between groups of island and coastal populations (FCT=0.1225).
The Bayesian assignment tests implemented in STRUCTURE (Pritchard et al., 2000) corroborated these high levels of inter-population differentiation, with little admixture among demes (Figure 1). Examining the likelihood of STRUCTURE assignments at various levels of K, we found that the model likelihood values plateau at a value of K between 6 and 10. Closer examination of assignments in that range indicated that the 17 sampled populations are best represented by eight genetic demes (Figure 1). Beyond K=8, STRUCTURE creates additional, highly admixed demes within sampled population, rather than identifying groups corresponding to specific localities or groups of localities. Overall, the eight genetic demes detected by STRUCTURE follow a geographic trend, with nearby populations often falling into the same genetic deme. Four demes were comprised of (1) the two northernmost coastal populations (KM3 and WL1), (2) two northern islands (COS and RED), (3) one northern island (PRU) and two central coastal populations (ITA and SUN), and (4) four southern islands (IB, TTQ, GAT and COS) and one southern coastal population (UNA). Four populations formed their own genetic demes, two of which were islands (PPQ and TAM) and two coastal (WL2 and JUR).
Island population sizes, migration rates and divergence times
Maximum likelihood estimates of current island population sizes varied greatly, ranging from 270 for PPQ, to 25 000 for IB (Table 2). However, we found no correlation between island area and relative population size (ANOVA; F1,7=0.1766; P=0.6890) or distance from coast and population size (ANOVA; F1,7=2.8772; P=0.1408). Likelihood estimates of ancestral population sizes for all island/coastal comparisons showed that all island populations diverged from larger ancestral populations (Table 2; IM tests of parameter differences; P-values<0.0001).
Maximum likelihood estimates of population migration rates also showed great variance, ranging from a rate of 0.038 from PRU to WL1, to a rate of 0.80 from SUN to ITA, with some populations showing levels of migration that could not be distinguished from zero (Table 3). IM showed no significant asymmetry in migration between coastal and island populations. Island populations had nonzero estimates of migration from coasts for the northern (WL1 with RED, PRU), central (SUN with TAM) and southern populations (UNA with IB, GAT, COS) (Table 3). Coastal populations also had nonzero estimates of migrations from northern islands (COU, PPQ, RED with WL1), central islands (TAM with SUN) and southern islands (TTQ, GAT, COS with UNA) (Table 3). Surprisingly, we found only two instances of nonzero estimates of migration among neighboring coastal populations (ITA with SUN and UNA with JUR), with all pairs of neighboring coastal populations showing estimates that could not be distinguished from zero in at least one direction (Table 3). Thus, as in the case of genetic diversity, there is no significant geographic trend in migration rates or a significant difference between coastal/coastal and coastal/island migration rates. We found no relationship between population migration rates into islands (2N2m2) and island area (ANOVA; F1,7=0.0812; P=0.7853) or island distance from coast (ANOVA; F1,7=0.0575; P=0.8185). Thus, island population size and migration rates of T. taophora are unaffected by the area of the island or its distance from the nearest coast. Finally, maximum likelihood estimates of time since divergence showed no significant differences between coastal/coastal comparisons and coastal/island comparisons, evidenced by largely overlapping ninety percent confidence intervals (Table 3). However, our data revealed a geographic trend in divergence times, with northern and central populations generally showing shorter times since divergence than southern populations (Table 3).
Discussion
Our analysis of genetic variability in island and coastal populations of Thoropa taophora indicates that island biogeography predictions are only partially applicable to recently isolated continental islands. Specifically, we found that island populations of T. taophora are significantly less genetically diverse than coastal populations, as would be predicted by isolation and drift in small populations. However, we detected no effect of island area or distance from coast on degree of genetic diversity, indicating that the processes that govern genetic diversity on continental islands are not at equilibrium. We discuss two processes specific to continental islands that could lead to our results: (1) the different genetic signatures expected in vicariance vs founding events, and (2) the effects of continued baseline levels of migration between coastal and island populations.
The genetic signature of vicariance in continental island formation
Reduced genetic diversity on oceanic islands is expected because island populations are typically founded by a few migratory individuals, resulting in extreme genetic bottlenecks (James, 1971; Clegg et al., 2002b; Bell et al., 2015b). However, genetic diversity of continental island populations, that were once continuous with the coast, is determined by vicariance (Hedges, 1996). Thus, the degree of population genetic loss will depend on the size of the populations remaining on newly formed continental islands. We found a significant reduction in genetic diversity in island populations of T. taophora; thus, genetic drift has in fact reduced the diversity of island populations since they split from the mainland. However, we found no effect of island area on the genetic diversity of island populations. One possible explanation for this may be the small size of our focal species; for smaller animals the availability of suitable habitat may be more important than the total size of the island. In this way, continental islands are comparable to habitat fragments, where the effects of genetic drift and population size depend on body size with larger species suffering greater effects of drift (Frankham, 1996). Thus, unlike in oceanic islands, the lack of a significant relationship between continental island area and the severity of genetic drift is likely explained in part by the body size of the species and the size of the population left by vicariant events.
Continued migration to and from continental island populations
Amphibians are generally poor dispersers because of physiological constraints, such as susceptibility to desiccation (Duellman and Trueb, 1994), and because they often have low vagility (Berven and Grudzien, 1990), and high site fidelity (Blaustein et al., 1994). Saltwater does not represent an absolute barrier to amphibian dispersal (Seppa and Laurila, 1999; Vences et al., 2003; Measey et al., 2007; Bell et al., 2015a), but dispersal ability over marine waters likely varies among amphibian species based on their natural histories and physiological tolerances to salinity. Thoropa possesses unusually high tolerance to salinity, yet island populations do show the genetic signature of reduced gene flow (Bokermann, 1965; Sazima, 1971; Abe and Bicudo, 1991; Brasileiro et al., 2010). Pairwise FST analyses indicate that saltwater has a significant role in the genetic differentiation of Thoropa taophora, with pairwise comparisons of coastal/island and island/island populations showing significantly larger FST values than coastal/coastal population comparisons (Figure 3). However, maximum likelihood estimates of migration rates calculated in IM (Hey and Nielsen, 2004) showed overall low migration rates among all types of populations and evidence of non-zero migration in many of the coastal and island populations. Surprisingly, we found nonzero estimates of migration between coastal and island populations in both directions (to and from the mainland populations). This is unexpected because mainland populations typically serve as source populations for less diverse island populations (James, 1971; Clegg et al., 2002b). However, our data show that connectivity among continental islands and the mainland is higher, perhaps due to the proximity between them, and the similarity of habitat across the region. Although the mechanism of dispersal in unknown, this species’ ability to tolerate saltwater suggests that individuals may be washed to islands, especially during storms. Alternatively, because these islands are subject to a high degree of human visitation, individuals could be intentionally or accidentally transported from mainland to island populations. If dispersal to islands is occurring at low levels but continuously, this reduces the effects of island distance on genetic diversity, because all of the islands in our study are exchanging migrants with each other or with coastal populations. Thus, the distances of continental islands from nearest coastal populations are likely less important in shaping the genetic diversity on islands whose distance from the coast fall within the dispersal ranges of species and/or when the species have high tolerances for salinity.
Population structure and implications for diversification
We estimated migration rates of effectively zero among three coastal population pairs that are geographically close (7–12 km apart). Combined with high levels of population structure, low levels of admixture, and generally low migration rates, our data suggest that populations of T. taophora are exchanging few genes in general, even in coastal populations inhabiting continuous habitats.
Studies of the natural history of this genus suggest several aspects of its life history that could contribute to the observed high levels of genetic structure and low levels of migration. Thoropa taophora prefers stream and moist rocky outcroppings (Feio et al., 2006), and features of this habitat are essential to their reproduction because their tadpoles develop in freshwater seeps that flow over streamside rocks (Bokermann, 1965; Rocha et al., 2002). The patchy distribution of these habitats could play a role in structuring the genetic diversity of this species at fine scales on the landscape. In addition, Thoropa taophora exhibits a polygonous mating system in which related females share breeding sites (Muralidhar et al., 2013) and male Thoropa guard their tadpoles and aggressively defend the developing young from other males (Giaretta and Facure, 2004). This territorial behavior might contribute to increased philopatry and reduced dispersal (Greenwood, 1980; Clobert et al., 2009) and the kin-biased breeding structure likely reduces genetic diversity within population (Muralidhar et al., 2013). These traits, combined with the landscape complexity of the Atlantic Coastal Forest, may interact to limit interpopulation dispersal and increase drift, and thus promote differentiation even in continuous coastal habitats.
A recent large-scale biogeographic study of T. taophora and its sister species T. miliaris showed high levels of phylogenetic diversification in these species likely due to past climatic oscillations, topographic complexity and the heterogeneity of habitats in the Atlantic Coastal Forest (Fitzpatrick et al., 2009; Carnaval et al., 2014). That study found that populations in Juréia were genetically isolated and diverged early in the history of remaining T. taophora populations (Fitzpatrick et al., 2009). Our results corroborate that this region is highly differentiated and shows reduced genetic diversity, likely due to past isolation from other coastal populations (Marques et al., 2004). Our study underscores that the genetic structuring of mainland populations of T. taophora occurs at very small spatial scales, likely due to a combination of historical and ongoing processes. However, our estimates of population divergence times indicate that Northern island populations showed more recent divergence times from their nearest coastal populations than Southern populations, and Central and Southern populations tended to have larger estimates of population size. This is inconsistent with the trends reported by Fitzpatrick et al. (2009), which indicate a North to South population expansion, with Southern populations experiencing more recent vicariant events. The different patterns detected by these two studies is likely due to scale of inference given by the different types of genetic data. Fitzpatrick et al. (2009) analyzed three mitochondrial loci (regions of cytochrome b, 16S, and NADH dehydrogenase subunit 2), whereas our study used nuclear microsatellite loci. Microsatellite loci are among the most rapidly evolving nuclear loci and thus our study may give insight on more recent population level trends.
Continental islands as analogs for anthropogenic habitat fragments
Our finding that continental island populations of T. taophora are affected by vicariance bottlenecks and experience low, but not negligible migration from mainland populations indicates that these islands might be analogous to habitat fragments in an anthropogenically-modified, but traversable matrix. Many studies have attempted to predict the genetic diversity and community composition of species remaining in habitat fragments using island biogeography theory (Simberloff and Abele, 1976; Wilcox and Murphy, 1985; Debinski and Holt, 2000; Lomilino and Perault, 2001). As in continental island taxa, species in fragmented habitat show a high degree of variance in how well they fit classical island biogeography theory (Debinski and Holt, 2000). Thus, the demographic processes contributing to the diversity of T. taophora may be best understood by examining the history of habitat fragmentation, and the degree of bottleneck occurring during that process, and assessing the habitat types that pose the most significant barrier to dispersal in those systems.
The observed pattern of reduced genetic diversity in island populations demonstrates the importance of barriers to dispersal for the persistence of amphibian species in habitat fragments. Thoropa taophora is one of several species endemic to the highly threatened biome of the Brazilian Atlantic Coastal Forest (Morellato and Haddad, 2000; Haddad et al., 2013). Our findings of low overall genetic diversity in many populations, combined with low levels of gene flow and high levels of genetic differentiation, indicate that this species is experiencing ongoing threats to its genetic diversity. To best preserve this, as well as the many other threatened endemics in the Atlantic Coastal Forest, it is important to understand the current levels of genetic diversity and areas that are suffering from the greatest reduction in gene flow due to habitat destruction. Our study indicates that many of the Northern populations have low effective population sizes and experience less gene flow. Thus, these populations may indicate the greatest conservation concern for this species, as well as other species that share this habitat. Overall, our study emphasizes the importance of examining past geologic factors and current demographic processes (such as barriers to dispersal) in determining diversity patterns of Atlantic Coastal Forest endemics.
Data archiving
Microsatellite data are available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.7p9v1.
References
Abbott I, Grant P . (1976). Nonequilibrial bird faunas on islands. Am Nat 110: 507–528.
Abe A, Bicudo J . (1991). Adaptations to salinity and osmoregulation in the frog Thoropa miliaris (Amphibia, Leptodactylidae). Zool Anz 227: 313–318.
Bell RC, Brasileiro CA, Haddad CFB, Zamudio KR . (2012). Evolutionary history of Scinax treefrogs on land-bridge islands in south-eastern Brazil. J Biogeogr 39: 1733–1742.
Bell RC, Drewes RC, Channing A, Václav G, Kielgast J, Lötters S, Stuart BL, Zamudio KR . (2015a). Over-seas dispersal of Hyperolius reedfrogs from Central Africa to the oceanic islands of São Tomé and Príncipe. J Biogeogr 42: 65–75.
Bell RC, Drewes RC, Zamudio KR . (2015b). Reed frog diversification in the Gulf of Guinea: overseas dispersal, the progression rule, and in situ speciation. Evolution doi:10.1111/evo.12623.
Berven K, Grudzien T . (1990). Dispersal in the wood frog (Rana sylvatica – Implications for genetic population-structure. Evolution 44: 2047–2056.
Bittkau C, Comes HP . (2005). Evolutionary processes in a continental island system: molecular phylogeography of the Aegean Nigella arvensis alliance (Ranunculaceae) inferred from chloroplast DNA. Mol Ecol 14: 4065–4083.
Blaustein A, Wake D, Sousa W . (1994). Amphibian declines – Judging stability, persistence, and susceptibility of populations to local and global extinctions. Conserv Biol 8: 60–71.
Bokermann W . (1965). Notas sobre as espécies de Thoropa Fitzinger (Amphibia, Leptodactylidae). Anais da Academia Brasileira de Ciências 37: 525–537.
Brasileiro CA, Martins M, Sazima I . (2010). Feeding ecology of Thoropa taophora (Anura: Cycloramphidae) on a rocky seashore in Southeastern Brazil. S Amer J Herpetol 5: 181–188.
Brown R, Guttman S . (2002). Phylogenetic systematics of the Rana signata complex of Philippine and Bornean stream frogs: reconsideration of Huxley's modification of Wallace's Line at the Oriental-Australian faunal zone interface. Biol J Linn Soc 76: 393–461.
Carlsson J . (2008). Effects of microsatellite null alleles on assignment testing. J Hered 99: 616–623.
Carnaval AC, Waltari E, Rodrigues MT, Rosauer D, VanDerWal J, Damasceno R et al. (2014). Prediction of phylogeographic endemism in an environmentally complex biome. Proc R Soc B 281: 20141461.
Calsbeek R, Smith T . (2003). Ocean currents mediate evolution in island lizards. Nature 426: 552–555.
Clegg S, Degnan S, Moritz C, Estoup A, Kikkawa J, Owens I . (2002a). Microevolution in island forms: The roles of drift and directional selection in morphological divergence of a passerine bird. Evolution 56: 2090–2099.
Clegg S, Degnan S, Kikkawa J, Moritz C, Estoup A, Owens I . (2002b). Genetic consequences of sequential founder events by an island-colonizing bird. Proc Natl Acad Sci USA 99: 8127–8132.
Clegg SM, Phillimore AB . (2010). The influence of gene flow and drift on genetic and phenotypic divergence in two species of Zosterops in Vanuatu. Philos Trans R Soc Lond B Biol Sci 365: 1077–1092.
Clobert J, Galliard J, Cote J, Meylan S, Massot M . (2009). Informed dispersal, heterogeneity in animal dispersal syndromes and the dynamics of spatially structured populations. Ecol Lett 12: 197–209.
Debinski D, Holt R . (2000). A survey and overview of habitat fragmentation experiments. Conserv Biol 14: 342–355.
Duellman W, Trueb L . (1994) Biology of Amphibians. The Johns Hopkins University Press: Baltimore, Maryland.
Duryea MC, Zamudio KR, Brasileiro CA . (2008). Characterization of microsatellite markers for Thoropa taophora (Anura, Cycloramphidae), a frog endemic to the Brazilian Atlantic Rain Forest. Mol Ecol Res 8: 663–665.
Ellegren H. . (2000). Microsatellite mutations in the germline: implications for evolutionary inference. Trends Genet 16: 551–558.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14: 2611–2620.
Evans B, Brown R, McGuire J, Supriatna J, Andayani N, Diesmos A, Iskandar D, Melnick D, Cannatella D . (2003). Phylogenetics of fanged frogs: Testing biogeographical hypotheses at the interface of the Asian and Australian faunal zones. Syst Biol 52: 794–819.
Excoffier L, Laval G, Schneider S . (2005). Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol Bioinform 1: 47–50.
Feio R, Napoli M, Caramaschi U . (2006). Taxonomic consideration of Thoropa miliaris (Spix, 1824) with revalidation and redescription of Thoropa taophora (Miranda-Ribeiro, 1923)(Amphibia, Anura, Leptodactylidae). Arquivos do Museu Nacional, Rio de Janeiro 64: 41–60.
Fitzpatrick SW, Brasileiro CA, Haddad CFB, Zamudio KR . (2009). Geographical variation in genetic structure of an Atlantic Coastal Forest frog reveals regional differences in habitat stability. Mol Ecol 18: 2877–2896.
Frankham R . (1997). Do island populations have less genetic variation than mainland populations? Heredity 78: 311–327.
Frankham R . (1996). Relationship of genetic variation to population size in wildlife. Conserv Biol 10: 1500–1508.
Giaretta A, Facure K . (2004). Reproductive ecology and behavior of Thoropa miliaris (Spix, 1824)(Anura, Leptodactylidae, Telmatobiinae). Biota Neotropica 4: 1–10.
Gill A . (1980). Evolutionary genetics of Channel Island Peromyscus. In: Power DM (ed) The California Islands: Proceedings of a Multidisciplinary Symposium. Haagen: Santa Barbara, CA, USA Vol 1, pp 719–743.
Goudet J . (1995). FSTAT (Version 1.2): A computer program to calculate F-statistics. J Hered 86: 485–486.
Grazziotin FG, Monzel M, Echeverrigaray S, Bonatto SL . (2006). Phylogeography of the Bothrops jararaca complex (Serpentes: Viperidae): past fragmentation and island colonization in the Brazilian Atlantic Forest. Mol Ecol 15: 3969–3982.
Greenwood PJ . (1980). Mating systems, philopatry and dispersal in birds and mammals. Anim Behav 28: 1140–1162.
Haddad CFB, Toledo LF, Prado CPA, Loebmann D, Gasparini JL, Sazima I . (2013). Guide to the Amphibians of the Atlantic Forest: Diversity and Biology. Editora Anolis Books, São Paulo.
Hedges S . (1996). Vicariance and dispersal in Caribbean biogeography. Herpetologica 52: 466–473.
Hey J, Nielsen R . (2004). Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D-persimilis. Genetics 167: 747–760.
Heyer WR, Rand AS, da Cruz CAG, Peixoto OL, Nelson CE . (1990). Frogs of Boracéia. Arquivos de Zoologia 31: 231–410.
Illera JC, Emerson BC, Richardson DS . (2007). Population history of Berthelot’s pipit: colonization, gene flow and morphological divergence in Macaronesia. Mol Ecol 16: 4599–4612.
Illera JC, Palmero AM, Laiolo P, Rodríguez F, Moreno ÁC, Navascués M . (2014). Genetic, morphological, and acoustic evidence reveals lack of diversification in the colonization process in an island bird. Evolution 68: 2259–2274.
Jaenike J . (1973). A steady state model of genetic polymorphism on islands. Am Nat 107: 793–795.
James J . (1971). The founder effect and response to artificial selection. Genet Res 12: 241–250.
Jordan MA, Snell HL . (2008). Historical fragmentation of islands and genetic drift in populations of Galapagos lava lizards (Microlophus albemarlensis complex). Mol Ecol 17: 1224–1237.
Kalinowski ST . (2005). HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5: 187–189.
Lomolino MV, Perault DR . (2001). Island biogeography and landscape ecology of mammals inhabiting fragmented, temperate rain forests. Global Ecol Biogeogr 10: 113–132.
Losos JB, Schluter D . (2000). Analysis of an evolutionary species-area relationship. Nature 408: 847–850.
MacArthur R, Wilson E . (1967) The Theory of Island Biogeography. Princeton University Press: Princeton, New Jersey.
Marques OAV, Sazima I, Marques OA, Duleba W . (2004). História natural dos répteis da estação ecológica Juréia-Itatins. In: Estação Ecológica Juréia-Itatins: Ambiente Físico, Flora e Fauna. Holos, Ribeirão Preto: São Paulo, Brazil. pp 257–277.
Mayr E . (1963) Animal Species and Evolution. Belknap: Cambridge, Massachusetts.
Measey GJ, Vences M, Drewes RC, Chiari Y, Melo M, Bourles B . (2007). Freshwater paths across the ocean: molecular phylogeny of the frog Ptychadena newtoni gives insights into amphibian colonization of oceanic islands. J Biogeogr 34: 7–20.
Meirmans PG, Van Tienderen PH . (2004). GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4: 792–794.
Morellato LPC, Haddad CFB . (2000). Introduction: The Brazilian Atlantic Forest. Biotropica 32: 786–792.
Muralidhar P, de Sá FP, Haddad CFB, Zamudio KR . (2013). Kin-bias, breeding site selection and female fitness in a cannibalistic Neotropical frog. Mol Ecol 23: 453–463.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
R Core Team. (2014). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org/.
Richards CM, Moore WS . (1996). A phylogeny for the African treefrog family hyperoliidae based on mitochondrial rDNA. Mol Phylogenet Evol 5: 522–532.
Ricklefs RE, Bermingham E . (2004). History and the species-area relationship in lesser Antillean birds. Am Nat 163: 227–239.
Rocha CFD, Van Sluys M, Bergallo HG, Alves MAS . (2002). Microhabitat use and orientation to water flow direction by tadpoles of the leptodactylid frog Thoropa miliaris in southeastern Brazil. J Herpetol 36: 98–100.
Rosenberg NA . (2004). DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4: 137–138.
Rowe G, Beebee TJC . (2007). Defining population boundaries: use of three Bayesian approaches with microsatellite data from British natterjack toads (Bufo calamita. Mol Ecol 16: 785–796.
Sazima I . (1971). The occurrence of marine invertebrates in the stomach contents of the frog Thoropa miliaris. Ciência e Cultura 23: 647–648.
Seppa P, Laurila A . (1999). Genetic structure of island populations of the anurans Rana temporaria and Bufo bufo. Heredity 82: 309–317.
Simberloff DS, Abele LG . (1976). Island Biogeography Theory and Conservation Practice. Science 191: 285–286.
Suguio K, Martin L . (1978). Quaternary marine formations of the states of São Paulo and southern Rio de Janeiro. In: International Symposium on Coastal Evolution in the Quaternary. University of São Paulo: São Paulo, Brazil Vol 1, pp 55.
Suguio K, Angulo R, Corrêa I, Tomazelli L, Willwock J, Vital H . (2005). Paleoníveis do mar e paleolíneas de costa. In: Souza C, Suguio K, Oliveira A, Oliveira P (eds) Quaternário Do Brasil. Holos: Ribeirão Preto, São Paulo, Brazil.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P . (2004). Micro-Checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538.
Vellend M . (2003). Island biogeography of genes and species. Am Nat 162: 358–365.
Velo-Antón G, Zamudio KR, Cordero-Rivera A . (2012). Genetic drift and rapid evolution of viviparity in insular fire salamanders (Salamandra salamandra. Heredity 108: 410–418.
Vences M, Vieites DR, Glaw F, Brinkmann H, Kosuch J, Veith M, Meyer A . (2003). Multiple overseas dispersal in amphibians. Proc R Soc B 270: 2435–2442.
Whittaker RJ, Willis KJ, Field R . (2001). Scale and species richness: towards a general, hierarchical theory of species diversity. J Biogeogr 28: 453–470.
Wilcox BA, Murphy DD . (1985). Conservation strategy - the effects of fragmentation on extinction. Am Nat 125: 879–887.
Acknowledgements
This project was funded by a National Science Foundation Biotic Survey Grant (DEB—0542848), a Cornell University for DYO internship to MCD, and FAPESP and CAPES post-doctoral fellowships to CAB. We thank CFB Haddad, NL Hülle, E Lucas, M Martins, HM Oyamaguchi, RJ Sawaya and MTC Thomé for help with field collections. Samples were collected under permits 02027. 021071/03-04 issued by the Instituto Brasileiro do Meio Ambiente. We thank S. Bogdanowicz for help with microsatellite development; Jeff Piestrak and the Cornell Mann Library GIS consulting group for assistance with maps; and the Zamudio Lab for comments on earlier versions of the manuscript. Molecular data were collected in the Cornell Evolutionary Genetics Core Facility and analyzed at the Cornell Computational Biology Service Unit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

Cite this article
Duryea, M., Zamudio, K. & Brasileiro, C. Vicariance and marine migration in continental island populations of a frog endemic to the Atlantic Coastal forest. Heredity 115, 225–234 (2015). https://doi.org/10.1038/hdy.2015.31
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2015.31
