
Abstract
The relative importance of the processes that generate and maintain biodiversity is a major and controversial topic in evolutionary biology with large implications for conservation management. The Atlantic Forest of Brazil, one of the world’s richest biodiversity hot spots, is severely damaged by human activities. To formulate an efficient conservation policy, a good understanding of spatial and temporal biodiversity patterns and their underlying evolutionary mechanisms is required. With this aim, we performed a comprehensive phylogeographic study using a low-dispersal organism, the land planarian species Cephaloflexa bergi (Platyhelminthes, Tricladida). Analysing multi-locus DNA sequence variation under the Approximate Bayesian Computation framework, we evaluated two scenarios proposed to explain the diversity of Southern Atlantic Forest (SAF) region. We found that most sampled localities harbour high levels of genetic diversity, with lineages sharing common ancestors that predate the Pleistocene. Remarkably, we detected the molecular hallmark of the isolation-by-distance effect and little evidence of a recent colonization of SAF localities; nevertheless, some populations might result from very recent secondary contacts. We conclude that extant SAF biodiversity originated and has been shaped by complex interactions between ancient geological events and more recent evolutionary processes, whereas Pleistocene climate changes had a minor influence in generating present-day diversity. We also demonstrate that land planarians are an advantageous biological model for making phylogeographic and, particularly, fine-scale evolutionary inferences, and propose appropriate conservation policies.
Similar content being viewed by others


Introduction
The causes of the origin and maintenance of extant biodiversity are central but controversial topics in evolutionary biology. Two hypotheses have dominated the debate on the major factors driving the high diversity of the Neotropics (Rull, 2011). The so-called refuge hypothesis (Haffer, 1969) proposes the recurrent aridity–humidity cycles caused by Pleistocene glaciations as the main factor. However, this hypothesis is contradicted by a wealth of paleoclimatic evidence and molecular dating data (Rull, 2008; Hoorn et al., 2010) showing that extant biodiversity originated before the Pleistocene period. An alternative hypothesis asserts that the major diversification events occurred much earlier, during the Neogene period, and were mainly promoted by geological reorganizations (tectonic or paleogeographical) (Simpson, 1979), and were also affected by biotic factors (for example, soil adaptations, biotic interactions and dispersal abilities) throughout the Quaternary and Neogene periods (Antonelli and Sanmartín, 2011). The origin of extant biodiversity would therefore be the result of complex interactions among these processes that act at global or regional scales (Rull, 2011). Hence, to obtain a complete understanding of the origin and distribution of biodiversity, it is critical to characterize the particular geological and paleoclimatic history of the region of interest, and to determine the evolutionary processes that have shaped extant biodiversity.
The Atlantic Forest of Brazil (AF) is one of the world’s most important biodiversity hot spots (Myers et al., 2000) and, at the same time, one of the most threatened biomes on the planet. The area of the AF is currently severely reduced (11–16% of its original coverage remains; Ribeiro et al., 2009), and it is seriously affected by habitat fragmentation (Gascon et al., 2000). Even so, the remaining forest fragments still harbour a high number of endemic species. To preserve present-day biodiversity, several areas distributed across these fragments have been protected (conservation areas). Policy initiatives have been put forth to connect these areas with biological corridors with the aim of facilitating gene flow and, hence, restoring ecosystem functionality. However, an efficient and responsible conservation policy requires knowledge of current biodiversity patterns and, more importantly, a better understanding of their evolutionary history.
Recent surveys applying paleoclimate modelling (Carnaval and Moritz, 2008; Carnaval et al., 2009; Amaro et al., 2012), phylogeographic analyses (Fitzpatrick et al., 2009; Thomé et al., 2010; D’Horta et al., 2011) and endemism distribution patterns (Costa et al., 2000) have concluded that the origin of AF genetic diversity predates the Pleistocene epoch. However, these analyses provide contradictory evidence regarding the influence of the Pleistocene glacial cycles on the extant patterns of genetic and species diversity. Paleoclimate modelling and some genetic studies support the existence of the forest throughout the last glaciations in the northern part of the AF (NAF; Figure 1; Cabanne et al., 2008) and a recent recolonization of southern AF regions (SAF; Figure 1; Grazziotin et al., 2006; Cabanne et al., 2007; Carnaval and Moritz, 2008; Carnaval et al., 2009). Nevertheless, data on endemism and genetic diversity studies (Pinto-Da-Rocha et al., 2005; Thomé et al., 2010; Amaro et al., 2012) suggest that during the glaciations, SAF regions may have maintained considerable diversity in forest-dependent species. Many of these studies have focussed on organisms with a high-dispersal ability (basically vertebrates, Cabanne et al., 2008; D’Horta et al., 2011) that may hinder making fine-scale predictions. Specialized and low-dispersal organisms can preserve the genetic signature of the past climate and geological events in small geographical areas (Hugall et al., 2002; Sunnucks et al., 2006; Marske et al., 2011). Thus, using a low-dispersal organism maximizes the probability, given comparable sampling designs, of detecting a finer-scale genetic structure within the AF relative to other studies using higher-dispersal co-distributed organisms.

Geographical distribution of the sampled populations across the SAF region (detailed in Supplementary Table S1). Present-day rain forest coverage is indicated by patched areas. Squares indicate the localities where C. bergi was not found. Lines divide the area of study in CAF, N-SAF, C-SAF and S-SAF.
Land planarians (Platyhelminthes, Tricladida, Geoplanidae) are particularly appropriate for fine geographical scale genetic studies (Sunnucks et al., 2006; Alvarez-Presas et al., 2011, 2012) because of their poor vagility and low capacity to prevent water loss, thus being strongly dependent on environmental moisture (Froehlich, 1955; McDonald and Jones, 2007). They seemingly withstand this limitation through behavioural strategies, such as hiding in damp soil refuges in the forest during the day and becoming active during the night. Although a few species are well adapted to open and human-transformed lands, most Neotropical species, including our focal species, Cephaloflexa bergi (Graff, 1899), are restricted to humid forest areas, chiefly natural forests (Carbayo et al., 2002). These animals are hermaphrodites with crossed fertilization (Winsor et al., 2004). Although for freshwater planarians some cases of asexuality are known, either by parthenogenesis or by fission, it is likely not the case in terrestrial planarians as we have never found worms showing the typical clear nonpigmented body end following fission, except asymmetrical marks resulting from accidental damage.
This study aims to contribute to the understanding of the processes underlying the origin and distribution of the biodiversity of the Brazilian Atlantic Forest and to gain insights into the discrepancies regarding the influence of Pleistocene glacial cycles on the extant patterns of biodiversity. To achieve these objectives, we focussed on C. bergi that we expect to mirror the forest distribution and to potentially be a good model to study the evolution of SAF biodiversity. We have conducted a comprehensive statistical phylogeographic analysis including several individuals per population that is fundamental for contrasting evolutionary scenarios using rigorous statistical methods based on the coalescent theory (Bertorelle et al., 2010). Specifically, we expected to find (1) intrapopulation nucleotide variation at or close to the mutational equilibrium with substantial interpopulation genetic structure in stable areas; and (2) molecular hallmarks of a recent population bottleneck and an absence of genetic patterns of isolation by distance in unstable areas. To address these questions, we simulated explicit demographic models representing scenarios of stability or recolonization of SAF areas with respect to the last glacial maximum (LGM; Carnaval et al., 2009). We evaluate the support of the data by the Approximate Bayesian Computation (ABC) approach that allows direct comparison of competing evolutionary models under a Bayesian framework. Overall, our results indicate that the origin of C. bergi diversity is ancient and that most SAF areas could have maintained high levels of diversity through the Pleistocene, probably including the southernmost areas. Our results suggest that extant patterns of genetic diversity in this species have been shaped by the interaction of climate-driven evolutionary processes and ancient geological events.
Materials and methods
Details for all the analyses are given in the Supplementary Methods.
Study area
Sampling was performed in 13 conservation areas within the Atlantic rainforest in Brazil (Figure 1 and Supplementary Table S1A). This rainforest, boasting different physiognomies, is an area varying from the coast up to 300 km inland, where rainfall is originated from oceanic winds and seaside mountain ranges (Oliveira-Filho and Fontes, 2000). Samples from each conservation area were treated as a population under the criteria of geographic proximity (the geographic distance among sampling sites within a conservation area were <10 km); moreover, we checked for genetic differentiation among individuals from different sampling sites within a conservation area and found no significant differences (on basis to the Snn statistic; see Supplementary Methods). We collected a total of 213 specimens of C. bergi that were cut into two pieces; one part was fixed in absolute ethanol and the other in 10% formalin.
Distribution modelling
We modelled the distribution of C. bergi for the current and LGM (21 kya) climatic periods using the localities sampled in this study (23 points) and the coordinates of the few localities cited in the literature (9 points) (Carbayo and Froehlich, 2008). As input data for the distribution models, we employed the same seven climatic variables as in other studies in the same region (Carnaval et al., 2009; Amaro et al., 2012); that is, annual mean temperature, temperature seasonality, mean temperature of warmest quarter, mean temperature of coldest quarter, annual precipitation, precipitation of wettest quarter and precipitation of driest quarter; information downloaded from the WORLDCLIM 1.4 database (http://www.worldclim.org/) (Hijmans et al., 2005). We selected these variables because the ecological requirements of terrestrial planarians, as related to the climatic variables, are presumably similar to those of the frogs used in previous analyses (chiefly high dependence on humidity). Climatic retrojections were performed using the Community Climate System Model (CCSM) and the Model for Interdisciplinary Research On Climate (MIROC). We used 75% of the localities in each replicate to train the model and 25% to test it.
DNA extraction, gene amplification and sequencing
We amplified and sequenced ∼800 bp of the mitochondrial gene cytochrome oxidase I (COI) and ∼500 bp of the nuclear ribosomal internal transcribed spacer-1 (ITS-1) as described in Alvarez-Presas et al. (2011).
Phylogenetic analysis
After the alignment, we determined the nucleotide substitution model that best fits the data applying the Akaike information criterion; the model obtained was GTR+I+G for both genes. We used Maximum Likelihood and Bayesian Inference methods to estimate phylogenetic relationships independently for COI and ITS-1 data sets. Outgroup species selection was based on a phylogenetic study of Geoplaninae (Carbayo et al., 2013).
We estimated the divergence time of extant lineages under Bayesian inference using COI sequence information. We used the uncorrelated lognormal relaxed clock model with a mean substitution rate of 0.017 nucleotide substitutions per site and per million years (Solà et al., 2013).
Population genetics and ABC-based analyses
Intra- and inter-population variation estimates not included in the ABC analyses were calculated in DnaSP (Librado and Rozas, 2009) by excluding ambiguous positions (that is, using the complete deletion option).
We conducted two different analyses (combining COI and ITS-1 data) in the ABC framework. First, to test whether a regional range expansion through a series of colonization events could explain current patterns of genetic variation, we applied the ABC–GLM (General Linear Model) method (Leuenberger and Wegmann, 2010) implemented in the ABCtoolbox package (Wegmann et al., 2010) to compare four models based on a general structural serial founder scheme (Supplementary Figure S1). In particular, we evaluated the hypothesis of the north to south migration proposed in previous studies (Cabanne et al., 2008; Carnaval et al., 2009). As alternatives, we evaluated three other scenarios (south to north; from the centre to south and to north; from north and south to the centre). These four scenarios tried to cover all possible cases, as biological differences among planarians and previously used model organisms (mainly small vertebrates) may result in different responses to the history of the region. The specific details about the ABC scheme, including prior distribution of model parameters, are specified in the Supplementary Methods.
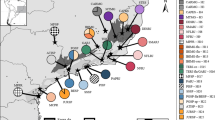
Second, we also conducted separate ABC analyses for each individual population (11+4 independent analyses; see below). We evaluated the support from the genetic data at each individual locality for the two demographic scenarios regarding the influence of the LGM proposed in the literature (Figure 2). In the scenario of stability (LGM Stability Model (LSM)) we simulated a population of arbitrary founder size, cNe (c can take values between 0 and 1), that originated Tc (120 000 to 5 000 000) years ago before the LGM. In the recolonization scenario (LGM Recolonization Model (LRM)) the locality is refounded Tb (0 to 21 000) years ago after the LGM by a small number of individuals (b ranges from 0 to 0.05). All prior distributions of the model parameters were set to uniform between the above-mentioned values. As we cannot use any of the sampling localities as the putative source for these models (see Discussion), we assumed that the source population (white populations in Figure 2) is at neutral equilibrium.

Demographic models used in the local ABC analyses. Panel left, LSM. Panel right, LRM. Current (sampled) and source (not sampled) populations are depicted in grey (light grey for LSM and dark grey for LRM) and white, respectively. Ne, effective population size of the source population. The letters c and b indicate the proportion of the source population that participates in the founding of current sampled SAF populations in the LSM and the LRM models, respectively. Tc and Tb denote the times of the founding events.
The prospective inspection of simulations results showed that some parameter values drawn from the prior, and therefore candidate to form part of the posterior distribution of interest, generate simulation replicates with no segregating site in some of the two markers. Given that in these cases some summary statistics cannot be computed (that is, they include a division by zero), we summarized our data sets by computing the statistic d—the difference between the average number of pairwise nucleotide differences between sequences in the sample (k) and the Watterson’s estimator of the population mutation rate θ (Tajima 1989), the haplotype diversity (H; Depaulis and Veuille, 1998) and the total number of segregating sites (S) in the sample. These three statistics are expected to be sensitive to changes in the effective population size (Ne), and to correlate with the severity of population size bottlenecks. We computed a vector of six summary statistics (the three above-mentioned statistics computed separately for each locus) in 1 000 000 simulated data sets under each of the two competing evolutionary models. The 5% of the simulations having the smallest Euclidean distances with the observed empirical data were retained to perform the model choice step. We applied the weighted multinomial logistic regression postsampling adjustment (Beaumont, 2008) implemented in the ‘abc’ package (Csilléry et al., 2012).
We validated our model selection procedure (independently for each population) by using pseudo-observed data sets (PODS) (Peter et al., 2010). In particular, for each demographic model (LSM and LRM) we generated 1000 PODS by sampling randomly from the prior distributions of the model parameters. We then computed the proportion of PODS where the true model was correctly assigned by our model choice (that is, the confusion matrix), and the mean posterior probabilities (PP) of the true model when the wrong model was chosen. As insufficient summary statistics can produce biased ABC-based PP estimates, we calibrated these probabilities by comparing the PP obtained in our ABC approach with those estimated in sets of 100 discrete bins of probability as in Peter et al. (2010).
Finally, we also validated the highest supported model in some localities. For that we recorded the fraction of retained simulations (the 5000 simulated data sets closest to the empirically observed data) that have a smaller or equal likelihood than the observed data under the GLM model (P-value of the model; Leuenberger and Wegmann, 2010; Wegmann et al., 2010). To test whether the estimation of model parameters is unbiased, we checked for the uniformity in the distribution of the posterior quantiles of true model parameters (Cook et al., 2006; Wegmann et al., 2010).
For the two ABC analyses the multilocus coalescent simulations were performed using the program mlcoalsim (version 1.98) (Ramos-Onsins and Mitchell-Olds, 2007), whereas the vector of the observed summary statistics was calculated with the program mstatspop (SE Ramos-Onsins; available at http://bioinformatics.cragenomica.es/numgenomics/people/sebas/software/software.html).
Results
Distribution of C. bergi and morphological identification
We found C. bergi at 11 of the 13 sampled localities (Figure 1 and Supplementary Table S1). This species was absent at 11-CJ and 34-FN. All of the 34 examined individuals exhibited the morphological and histological diagnostic features of C. bergi (see Carbayo and Leal-Zanchet, 2003), and these specimens constituted a monophyletic group in the phylogenetic analysis including other species. We obtained COI and ITS-1 DNA sequences from 213 and 130 individuals, respectively.
C. bergi distribution modelling
Both prediction models, run under current climatic conditions and projections for the LGM, provided areas under the receiver operating characteristic curve values of >0.9, which indicates that the models perform well (Supplementary Figure S2). The CCSM and MIROC predict that the AF coast, including the southernmost regions, presented climatic suitability for C. bergi during the LGM; the MIROC model predicts a very similar distribution 21 kya to that for present days. Nevertheless, as the prediction models are based on only a few data points, past projections might be not sufficiently accurate.
Phylogenetic relationships
The COI alignment resulted in an 822-bp matrix with no gaps and a total of 226 polymorphic sites (for C. bergi); for ITS-1 the final matrix after running GBlocks contained 451 bp, with 107 polymorphic sites for C. bergi. The list of C. bergi individuals and outgroups included in the analyses together with its GenBank Accession Numbers are provided in Supplementary Table S1. The COI-based tree topologies inferred through Maximum Likelihood and Bayesian Inference (Figure 3a and Supplementary Figure S3A) defines three well-differentiated phylogenetic groups that are associated with the geographical distribution of the C. bergi populations (designated N-SAF, C-SAF and S-SAF for the northern, central and southern South Atlantic Forest populations, respectively; Figure 1). Five populations are polyphyletic, exhibiting highly divergent lineages (01-AR, 19-PC, 25-SL, 28-SI and 31-ST), with two of them including individuals from two of the major clades (19-PC from C-SAF and N-SAF and 25-SL from S-SAF and C-SAF). Although in some of these populations the animals were collected in two or more sampling sites (Supplementary Table S1), the highly divergent genetic lineages (denoted as clade 1 or 2, Figure 3a) found within a population do not coincide with the sampling sites.

Maximum likelihood-based tree inferred from the COI (a) and ITS-1 (b) data sets. Bootstrap values (⩾50%) are shown above nodes. Monophyletic groups constituted by individuals of the same population have been collapsed (Supplementary Figure S3). (c) Neighbour-joining tree inferred from COI DXY values. The scale bars represent the number of substitutions per site.
The ITS-1 tree exhibits basically the same topology as that for COI (Figure 3b, Maximum Likelihood; Supplementary Figure S3B, BI). For both genes, the 01-AR population (within the Central Atlantic Forest area (CAF); Figure 1) is polyphyletic; one of the lineages is basal to the rest of the populations, whereas the other includes extremely long branches that are phylogenetically close to N-SAF or S-SAF clade lineages. Because long branches might generate artefactual clustering, we reanalysed the COI data after excluding the fastest evolving sites, that is, those belonging to the fifth category, as calculated with MEGA (http://www.megasoftware.net/) (Supplementary Figure S4). The analyses show that 01-AR is monophyletic, but its position in the tree is not resolved; the rest of the populations maintain their phylogenetic relationships, indicating that the anomalous behaviour of 01-AR is probably due to a Long Branch Attraction problem.
The analysis using BEAST (http://beast.bio.ed.ac.uk/) (Supplementary Figure S5) shows that the divergence times of the most recent common ancestor of the deepest lineages of polyphyletic populations are very old (∼4.5–7 Mya). In the case of the 19-PC and 25-SL, this timing is very close to the split of the three major geographic clades (N-SAF, C-SAF and S-SAF; ∼7 Mya, 95% highest posterior density 5.8–8.5). Therefore, the origin of the extant genetic variation likely dates back to the Miocene epoch.
Intrapopulation DNA sequence variation and neutrality tests
We found that the levels of nucleotide diversity (π) for both the COI and ITS-1 markers (Table 1) were high compared with those estimated for other planarian species (Sunnucks et al., 2006; Álvarez-Presas et al., 2012) and even for other organisms (Goodall-Copestake et al., 2012). Many populations are far from the neutral equilibrium, and not all populations deviate in the same direction (Table 1 and Supplementary Table S2). For instance, several populations (01-AR, 19-PC and 31-ST) show significant positive Tajima’s D values for COI, whereas 28-SI exhibits significant negative values. A similar trend is also observed for the Fu’s FS and Ramos-Onsins and Rozas’s R2 test statistics at both markers, although the most significant cases are at the right tail. In agreement with the previous results, the within-population linkage disequilibrium, analysed using the ZnS and Wall’s Q statistics, is also far from neutral expectations. In fact, these deviations are found in polyphyletic populations, and reflect the strong structure found in these samples.
Interpopulation DNA divergence
As anticipated based on the inferred tree topology and the high-nucleotide diversity values, the interpopulation genetic distances were quite large (Supplementary Table S3); for instance, DXY reaches 0.107 for COI and 0.151 for ITS-1. Moreover, the surveyed populations were highly differentiated (Supplementary Tables S4 and S5), resulting in very low levels of gene flow (Nm=0.09 and Nm=0.06 for COI and ITS-1, respectively). We found in the DXY population-based dendrograms that geographically close populations are also genetically related, both for COI (Figure 3c) and for ITS-1 (Supplementary Figure S3C). More importantly, the N-SAF, C-SAF and S-SAF geographical areas are clearly separated in the trees. Unlike the Maximum Likelihood and Bayesian Inference phylogenetic trees based on individuals, the DXY population-based dendrograms (Figure 3c and Supplementary Figure S3C) display a close relationship between the N-SAF and C-SAF clades, although the internal branch connecting the two groups is short. This analysis also shows that the 01-AR population (CAF) is basal to the other C. bergi populations. The geographical and genetic distances between all population pairs are significantly correlated (Mantel test, r=0.527, P<0.001 for COI; r=0.426, P=0.003 for ITS-1; Supplementary Table S6).
ABC-based analyses: regional models
We applied the ABC approach to investigate the origin of the present-day geographic distribution of nucleotide diversity across the SAF. We first tested four putative regional scenarios based on serial population founder event models (Supplementary Figure S1). Model 1, which emulates a range expansion from the northernmost populations, shows the highest posterior probability (PP=0.427), although it is not more strongly supported than the other three models, and the P-value of the model under the GLM is very low (P-value=0.023) (Supplementary Table S7). Hence, these results should be interpreted with caution because the underlying models include a large number of parameters (that is, a large number of populations), and the modelled scenarios are likely too simple and ignore particular intrapopulation demographic histories.
ABC-based analyses: local models
Our validation analyses based on PODS confusion matrices (Supplementary Figure S6) demonstrate that our model choice procedure correctly identified the true model in the 80.7% of the cases (74% and 87.3% for LRM and LSM models, respectively). The average posterior probabilities supporting the model when the wrong model is chosen is relatively low in all cases (average PP of the wrong model, PP<0.300), indicating that the wrong model is rarely chosen with high probability. Moreover, calibration analysis show that although posterior probabilities from the ABC method are slightly biased towards one of the two models in some localities (Supplementary Figure S7), our model choice inference can be considered relatively unbiased in most cases (but see below). These results indicate that our model choice procedure is powerful and unbiased enough to distinguish between the LSM and the LRM scenarios with high reliability.
We found that in 5 of the 11 SAF localities, the LRM (that is, involving a bottleneck associated with a recent colonization after the LGM; Figure 2) is more supported by the data than the LSM (PPLRM>0.500: Figure 4a). Nevertheless, in three of these localities (01-AR, 19-PC and 31-ST), the P-value of the model under the GLM is very low (0.058, 0.021 and 0.065, respectively), suggesting that it is highly unlikely that LRM generated the observed data. In fact, phylogenetic trees and neutrality-based tests (Table 1) indicate that these populations are far from neutral equilibrium and suggest the existence of some genetic structure in the samples. To gain insights into the biological meaning of such genetic structure, we performed the ABC model choice separately in some genetic lineages (genetic clusters) of the polyphyletic localities. In particular, we reanalysed the data in three clusters (we reanalysed the data only in clades with n⩾4 in both markers; that is, clade 1 in 01-AR; clades 1 and 2 in 19-PC; Figure 3a). The results of these new analyses show that the LSM model is always the highest supported model with high posterior probabilities (Figure 4b); moreover, the likelihood of the observed data is high (P-value >0.500) in all cases. These results suggest that current coexistence in a single locality of two (or more) of such divergent genetic clades might reflect secondary (and recent) contacts of highly diverged populations that were stable during the Pleistocene epoch.

Posterior probabilities of the LSM (light grey) and the LRM (dark grey) models inferred in the ABC approach. (a) Analysis using all samples from each locality. (b) Analysis using highly diverged clades. Numbers on bars indicate the P-values of the data under the General Linear Model (GLM) estimated with the selected demographic model. Asterisks denote the localities where the data observed are highly unlikely even under the best-supported model (low P-values).
For the other two localities with high PP values of the LRM model (04-ED and 13-EC; Figure 4a) the likelihood of the observed data is very high (P-values of 0.886 and 0.973, respectively). Nevertheless, these two localities show the stronger bias in the ABC posterior probabilities, especially around the estimated values (Supplementary Figure S7). After calibrating these probabilities empirically, the LRM is no longer the highest supported model in 04-ED (Empirical PPLRM_04-ED<0.500). This is not the case of 13-EC, where after calibrating the probability the data from this locality could also favour the LRM model (PP>0.700). In fact, this probability is still higher than the average probability favouring LRM model in PODS generated under LRM (average PP=0.678).
For the rest of localities, we found that the LSM model is strongly supported in 07-SO (PPLRM=0.915) and 10-SB (PPLRM=0.795). In 16-BB, 22-PI, 25-SL and 28-SI, the LSM model is also the best model but with PP only slightly higher than the LRM (PPLSM<0.60; Figure 4a). The result of the ABC model choice for these localities is therefore inconclusive. Yet, the likelihood of the data under the selected model in three of them is sensibly lower than that of the other localities (16-BB, P-value=0.167, 25-SL, P-value=0.101 and 28-SI, P-value=0.005). The case of 28-SI is especially noteworthy. Like 01-AR, 19-PC and 31-ST, the samples from this locality are polyphyletic, and show signals of intralocality genetic structure. The reanalysis of the data in one of the divergent clades shows that the LSM model remains to be the best model, with even higher posterior probabilities than that estimated using all the specimens from this locality (Figure 4); nevertheless, the likelihood of the observed data is still low that indicate that the LSM model is not a good model to explain 28-SI data.
The visual inspection of the posterior estimates under the selected models reveals that the posterior densities of both the colonization time and the size of the founder population are too wide for both models (Supplementary Figure S8). A significant bias in the estimation of these posteriors could explain these results. Yet, the distribution of posterior quantiles of the true parameters indicates these estimates are not biased (Supplementary Figure S9), suggesting that the data do not provide sufficient information to obtain precise estimates of model parameters. Nevertheless, our main objective was not obtaining precise estimates of specific model parameters. Rather, our purpose was to test alternative demographic scenarios and to discriminate among probable and highly improbable models, and this we achieved with reasonable power and accuracy based on C. bergi data.
Discussion
There is an increasing number of studies assessing the impact of the so-called refugia hypothesis using powerful molecular and statistical tools. Here, we contribute to these analyses using a low-mobility and highly forest-dependent organism. We carried out a broad sampling, including several individuals per locality (an average of 19 individuals and 20 polymorphic sites per population at the COI region; Table 1, upper), which allowed us to conduct a reasonably powerful statistical phylogeographic analysis.
C. bergi genetic diversity has an ancient origin
The deep nodes inferred in the phylogenetic trees (at the intra- and inter-population levels) and the high estimated nucleotide variation within and between populations indicate an old origin of C. bergi diversity. Our BEAST-based dating results (Supplementary Figure S5) reveal that the origin of within-species diversity (∼8.5 Mya, 95% highest posterior density 6.5–10.6) clearly predates the last glaciation period (Ledru et al., 2009); that is, the split between C. bergi and the outgroup species occurred in the Miocene epoch, but more importantly, many of the within-species splits also predate the early Pleistocene.
C. bergi genetic data do not support a recent colonization of the SAF
Our analyses do not support the hypothesis that C. bergi colonized the SAF region recently (after LGM) for the following reasons: (1) there is no evidence of the characteristic molecular fingerprint left by recent population expansions; (2) we showed that populations exhibit high levels of nucleotide diversity and deep coalescent times (until ∼8.5 Mya); (3) SAF localities present the characteristic isolation-by-distance signal that is not expected for a low-mobility organism in a recently colonized region; (4) there is no clear support for any of the four evaluated regional range expansion scenarios; and (5) the population genetic data either substantially support the LSM or are inconclusive with respect to the most probable model (Figure 5), but certainly not show a clear support for the LRM model (with the exception of 13-EC; see below).

Graphical representation of the local ABC results in a geographical context. Predicted forest-covered areas 21 000 years ago are indicated in light blue in the map (narrower definition of Carnaval and Moritz, 2008). The colour of the inner circle indicates the best-supported model (light grey, LSM; dark grey, LRM; diagonal lines pattern, LSM with PP<0.600). The colour of the outer circle represents the fit of the estimated GLM to the observed data (black, good fit. white, poor fit). In localities 01-AR, 19-PC and 28-SI, we only show results of the ABC analysis in separated clades (small circles). The results of the locality 04-ED are based on the calibrated posterior probability. A full color version of this figure is available at the Heredity journal online.
Overall, our genetic diversity based results clearly indicate the existence of C. bergi populations during the LGM in the most part (at least from Espírito Santo to São Paulo) of the studied SAF region, with the exception of the region occupied by locality 13-EC. Although our paleoclimatic model-based predictions for C. bergi might not be sufficiently accurate, they also support the preceding statement. The findings of our survey are also consistent with reports detecting a great number of endemic species (Costa et al., 2000; Pinto-Da-Rocha et al., 2005) and a high genetic diversity in southern regions (Cabanne et al., 2008). Although our results also agree with recent studies in Amphibia showing stable areas throughout the entire São Paulo coastal region (Amaro et al., 2012), we detect substantial genetic variation even in states south of São Paulo. The southernmost localities, however, exhibit a different characteristic. Independently of whether LSM or LRM models is more likely, none of them reproduces sufficiently well the observed pattern of genetic variation (the likelihood of the data under both models is very low; Figure 4), specially for 28-SI (and the clade 28-SI_1) and for 31-S. Therefore, in spite that there is no support for a simple (and recent) recolonization of these areas, we cannot discard the possibility of more complex recolonization scenarios.
The diversity of the SAF region was shaped by complex interactions between geological and demographic processes
The origin of the three major genetic clades that delimit three SAF regions (clearly uncovered by the DXY analysis as N-SAF, C-SAF and S-SAF; Figure 3c) and of 01-AR (within the CAF area) can be explained by the existence of major geological or geographical barriers. The Guapiara lineament and the Paranapanema River delimit an area between the C-SAF and S-SAF, and the Upper Rio São Francisco crustal discontinuity and the Campo do Meio fault zone delimit an area between the C-SAF and N-SAF (Thomé et al., 2010). Moreover, the subdivision of C-SAF into two groups seen in all phylogenetic analyses (16-BB and 19-PC vs 13-EC and 22-PI) could be related to the formation of the Paraíba do Sul River Valley (from 15 to 1 Mya; Petri and Fúlfaro, 1983; Silva and Straube, 1996; D’Horta et al., 2011), a process that gave rise to two of the major AF mountain ranges, the Serra do Mar and Serra da Mantiqueira. Notably, as expected for a major geological event, this pattern is also observed in other species, such as birds (Cabanne et al., 2007; D’Horta et al., 2011) and harvestmen (Pinto-Da-Rocha et al., 2005). The basal position of population 01-AR (Figure 3c and Supplementary Figure S3C) suggests that a more ancient physical barrier, the Paraiba do Sul basin, prevented the gene flow between the CAF and SAF.
In the N-SAF and C-SAF, most of the populations exhibit high levels of nucleotide diversity, showing a pattern compatible with the standard neutral equilibrium and the LSM scenario. Therefore, these areas should be climatically stable regions for C. bergi (in agreement with predictions of paleoclimate models for forests, frogs and even C. bergi), and thus they were subjected to little or no influence of recent Pleistocene glacial activity. The Cantareira (13-EC) population might represent a particular case. This park houses a secondary forest (of very recent origin) as a consequence of the modifications it has probably suffered as a side effect of being close to the ever-increasing urbanization and road building in the metropolitan region of São Paulo—inhabited by over 20 million people. This feature could explain why the recent bottleneck hallmark has been clearly detected only in this locality.
Our results support that climate changes could have played an important role in shaping, rather than originating, the genetic diversity observed in the SAF region. Indeed, the local ABC analyses reveal that at some localities the populations found nowadays might be formed by a merge of old populations (secondary and recent contacts) that could have stayed stable during the Pleistocene. This situation could be generated if genetically differentiated populations of land planarians (which might result from ancient processes such as isolation by distance or due to ecological or geographical barriers) existed in this region long before the LGM. More recently, these genetically differentiated populations might have come into contact driven by Pleistocene climate changes. Indeed, the most divergent lineages within the most structured populations are formed by the same individuals in both markers; that is, the timing of the secondary contact is so recent that recombination between the mitochondrial and nuclear regions has not had the opportunity to occur yet. This hypothesis might explain the ABC results of 01-AR and 19-PC. The alternating cycles of cold and warm phases promoted changes in the forest composition (Oliveira-Filho and Fontes, 2000; Grazziotin et al., 2006) that could generate new contact regions. Moreover, sea-level changes associated with Pleistocene ice mass retractions and expansions (Suguio and Martin, 1978) may have resulted in repeated cycles of isolation and merging of populations on the coast, which may explain the presence in the 19-PC population of lineages related to northern populations. Our analyses uncover another putative merging between populations situated on both sides of the ancient geological barriers that originated the three main clades. Some 25-SL lineages share a common ancestor with the 16-BB and 19-PC (C-SAF) populations (Figure 3a); this feature may be explained by the recent arrival of coastal C-SAF populations crossing the old barrier and coming into contact with ancient S-SAF lineages.
The coexistence of such high-diverged clades within a single locality might in fact represent some genetically isolated lineages from unrecognized cryptic species (species generated by putative old reproductive isolation barriers, in the absence of gene flow). Our morphological identification results, however, do not support this hypothesis; indeed, all specimens unambiguously matched the diagnostic characters described for the species, whereas the observed slight variations in dorsal colour patterns and length of copulatory apparatuses are not related either to geographic areas or to particular populations or lineages.
Conservation policy outcome
As a consequence of the poor vagility and high sensitivity to environmental modifications, the genetic structure of the populations of C. bergi might mirror the biota’s history. The Serra do Mar biological corridor was once a continuous forest landscape. With the aim of conserving and restoring the Atlantic forest, the protection of some SAF areas has been prioritized on the basis of the occurrence of certain target species and landscape structural features (Joly et al., 2010). From the results of our ongoing study, if confirmed by further surveys, three priority actions could complement these current scientifically based institutional initiatives by providing new executive insights for the Serra do Mar corridor. First, the reconnection of the Parque Estadual do Desengano (04-ED) to the Parque Nacional da Serra dos Órgãos (07-SO), the most genetically closely related populations, but the former has been isolated from protected areas through human-caused landscape transformation (Rocha et al., 2003). Second, to reconnect the Parque Estadual da Cantareira (13-EC) and the Parque Estadual Intervales (22-PI). Furthermore, this action could likely help to alleviate human-induced threatens to 13-EC. Lastly, the boundary of the Serra do Mar corridor might be extended to the South to embrace Parque Nacional da Serra de Itajaí (28-SI) and Parque Estadual da Serra do Tabuleiro (31-ST), as these two areas plus Parque Nacional Saint Hilaire-Lange (25-SL) constitute an evolutionarily geographical unit (Figure 1).
In conclusion, this study uncovers a complex scenario in which the extant patterns of biodiversity in the SAF resulted from convoluted interactions among ancient geological events that shaped the distribution of lineages, and more recent climate driving evolutionary processes. It confirms the utility of land planarians as a model for phylogeographic analyses, allowing levels of resolution that can improve those obtained using organisms with a higher mobility, and complementing the information provided by current paleoclimate models. This feature is particularly important for providing policy makers with the sound scientifically based technical information required to evaluate the effectiveness of future conservation strategies, such as the establishment of the most important areas to be protected before losing undocumented biological variability, or the design of effective biological corridors that could restore ecosystem functionality. In this context, good knowledge of spatial and temporal biodiversity patterns and the underlying evolutionary processes is critical.
Data archiving
COI and ITS-1 alignments available from the Dryad Digital Repository: doi:10.5061/dryad.t9584. Details regarding sampling site localities, individual samples and GenBank accession numbers are available in Supplementary Table S1.
References
Alvarez-Presas M, Carbayo F, Rozas J, Riutort M . (2011). Land planarians (Platyhelminthes) as a model organism for fine-scale phylogeographic studies: understanding patterns of biodiversity in the Brazilian Atlantic Forest hotspot. J Evol Biol 24: 887–896.
Álvarez-Presas M, Mateos E, Vila-Farré M, Sluys R, Riutort M . (2012). Evidence for the persistence of the land planarian species Microplana terrestris (Müller, 1774) (Platyhelminthes, Tricladida) in microrefugia during the Last Glacial Maximum in the northern section of the Iberian Peninsula. Mol Phyl Evol 64: 491–499.
Amaro RC, Rodrigues MT, Yonenaga-Yassuda Y, Carnaval AC . (2012). Demographic processes in the montane Atlantic rainforest: molecular and cytogenetic evidence from the endemic frog Proceratophrys boiei. Mol Phyl Evol 62: 880–888.
Antonelli A, Sanmartín I . (2011). Why are there so many plant species in the Neotropics? Taxon 60: 403–414.
Beaumont M . (2008) Joint determination of topology, divergence time, and immigration in population trees. McDonald Institute Monographs. Cambridge McDonald Institute for Archeological Research: UK. edn, pp 134–154.
Bertorelle G, Benazzo A, Mona S . (2010). ABC as a flexible framework to estimate demography over space and time: some cons, many pros. Mol Ecol 19: 2609–2625.
Cabanne GS, Santos FR, Miyaki CY . (2007). Phylogeography of Xiphorhynchus fuscus (Passeriformes, Dendrocolaptidae): vicariance and recent demographic expansion in southern Atlantic forest. Biol J Linn Soc 91: 73–84.
Cabanne GS, d’Horta FM, Sari EHR, Santos FR, Miyaki CY . (2008). Nuclear and mitochondrial phylogeography of the Atlantic forest endemic Xiphorhynchus fuscus (Aves: Dendrocolaptidae): biogeography and systematics implications. Mol Phyl Evol 49: 760–773.
Carbayo F, Leal-Zanchet AM, Vieira EM . (2002). Terrestrial flatworm (Platyhelminthes: Tricladida: Terricola) diversity versus man-induced disturbance in an ombrophilous forest in southern Brazil. Biodivers Conserv 11: 1091–1104.
Carbayo F, Leal-Zanchet AM . (2003). Two new genera of geoplaninid land planarians (Platyhelminthes: Tricladida: Terricola) of Brazil in the light of cephalic specialisations. Invertebr Syst 17: 449–468.
Carbayo F, Froehlich EM . (2008). Estado do conhecimento dos macroturbelários (Platyhelminthes) do Brasil. Biota Neotrop 8: 177–197.
Carbayo F, Álvarez-Presas M, Olivares CT, Marques FPL, Froelich EM, Riutort M . (2013). Molecular phylogeny of Geoplaninae (Platyhelminthes) challenges current classification: proposal of taxonomic actions. Zool Scr 42: 508–528.
Carnaval AC, Moritz C . (2008). Historical climate modelling predicts patterns of current biodiversity in the Brazilian Atlantic forest. J Biogeogr 35: 1187–1201.
Carnaval AC, Hickerson MJ, Haddad CFB, Rodrigues MT, Moritz C . (2009). Stability predicts genetic diversity in the Brazilian Atlantic forest hotspot. Science 323: 785–789.
Cook S, Gelman A, Rubin D . (2006). Validation of software for Bayesian models using posterior quantiles. J Comp Graph Stat 15: 675–692.
Costa LP, Leite YLR, Da Fonseca GAB, Da Fonseca MT . (2000). Biogeography of South American forest mammals: endemism and diversity in the Atlantic Forest. Biotropica 32: 872–881.
Csilléry K, François O, Blum MGB . (2012). abc: an R package for approximate Bayesian computation (ABC). Meth Ecol Evol 3: 475–479.
Depaulis F, Veuille M . (1998). Neutrality tests based on the distribution of haplotypes under an infinite-site model. Mol Biol Evol 15: 1788–1790.
D’Horta FM, Cabanne GS, Meyer D, Miyaki CY . (2011). The genetic effects of Late Quaternary climatic changes over a tropical latitudinal gradient: diversification of an Atlantic Forest passerine. Mol Ecol 20: 1923–1935.
Fitzpatrick SW, Brasileiro CA, Haddad CFB, Zamudio KR . (2009). Geographical variation in genetic structure of an Atlantic Coastal Forest frog reveals regional differences in habitat stability. Mol Ecol 18: 2877–2896.
Froehlich CG . (1955). On the biology of land planarians. Boletim da Facultade de Filosofia, Ciências e Letras da Universidade de Sâo Paulo 20: 263–271.
Gascon C, Williamson GB, Da Fonseca GAB . (2000). Receding forest edges and vanishing reserves. Science 288: 1356–1358.
Goodall-Copestake WP, Tarling GA, Murphy EJ . (2012). On the comparison of population-level estimates of haplotype and nucleotide diversity: a case study using the gene cox1 in animals. Heredity 109: 50–56.
Graff Lv . (1899). Monographie der Turbellarien II. Tricladida terricola (landplanarien). Engelmann: Leipzig.
Grazziotin FG, Monzel M, Echeverrigaray S, Bonatto SL . (2006). Phylogeography of the Bothrops jararaca complex (Serpentes: Viperidae): past fragmentation and island colonization in the Brazilian Atlantic Forest. Mol Ecol 15: 3969–3982.
Haffer J . (1969). Speciation in amazonian forest birds. Science 165: 131–137.
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A . (2005). Very high resolution interpolated climate surfaces for global land areas. Int J Clim 25: 1965–1978.
Hoorn C, Wesselingh FP, Ter Steege H, Bermudez MA, Mora A, Sevink J et al. (2010). Amazonia through time: Andean uplift, climate change, landscape evolution, and biodiversity. Science 330: 927–931.
Hugall A, Moritz C, Moussalli A, Stanisic J . (2002). Reconciling paleodistribution models and comparative phylogeography in the Wet Tropics rainforest land snail Gnarosophia bellendenkerensis (Brazier 1875). Proc Natl Acad Sci USA 99: 6112–6117.
Joly CA, Rodrigues RR, Metzger JP, Haddad CFB, Verdade LM, Oliveira MC et al. (2010). Biodiversity conservation research, training, and policy in São Paulo. Science 328: 1358–1359.
Ledru M, Mourguiart P, Riccomini C . (2009). Related changes in biodiversity, insolation and climate in the Atlantic rainforest since the last interglacial. Palaeogeogr Palaeoclimatol Palaeoecol 271: 140–152.
Leuenberger C, Wegmann D . (2010). Bayesian computation and model selection without likelihoods. Genetics 184: 243–252.
Librado P, Rozas J . (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Marske KA, Leschen RAB, Buckley TR . (2011). Reconciling phylogeography and ecological niche models for New Zealand beetles: looking beyond glacial refugia. Mol Phyl Evol 59: 89–102.
McDonald JC, Jones HD . (2007). Abundance, reproduction, and feeding of three species of British terrestrial planarians: observations over 4 years. J Nat Hist 41: 293–312.
Myers N, Mittermeler RA, Mittermeler CG, Da Fonseca GAB, Kent J . (2000). Biodiversity hotspots for conservation priorities. Nature 403: 853–858.
Oliveira-Filho AT, Fontes MAL . (2000). Patterns of floristic differentiation among atlantic forests in southeastern Brazil and the influence of climate. Biotropica 32: 793–810.
Peter BM, Wegman D, Excoffier L . (2010). Distinguishing between population bottleneck and population subdivision by a Bayesian model choice procedure. Mol Ecol 19: 4648–4660.
Petri S, Fúlfaro VJ . (1983) Geologia do Brasil—Fanerozoico. São Paulo: Editora da Universidade de São Paulo.
Pinto-Da-Rocha R, Da Silva MB, Bragagnolo C . (2005). Faunistic similarity and historic biogeography of the harvestmen of southern and southeastern Atlantic Rain Forest of Brazil. J Arachnol 33: 290–299.
Ramos-Onsins RE, Mitchell-Olds T . (2007). mlcoalsim: multilocus coalescent simulations. Evol Bioinform Online 2: 41–44.
Ribeiro MC, Metzger JP, Martensen AC, Ponzoni FJ, Hirota MM . (2009). The Brazilian Atlantic Forest: how much is left, and how is the remaining forest distributed? Implications for conservation. Biol Conserv 142: 1141–1153. 1141.
Rocha CFD, Bergallo HG, Alves MAS, Van Sluys M . (2003) A biodiversidade nos grandes remanescentes florestais do Estado do Rio de Janeiro e nas Restingas da Mata Atlântica. RiMa: São Carlos.
Rull V . (2008). Speciation timing and neotropical biodiversity: the Tertiary–Quaternary debate in the light of molecular phylogenetic evidence. Mol Ecol 17: 2722–2729.
Rull V . (2011). Neotropical biodiversity: timing and potential drivers. Trends Ecol Evol 26: 508–513.
Silva JMC, Straube FC . (1996). Systematics and biogeography of scaled woodcreepers (Aves: Dendrocolpatidae). Stud Neotrop Fauna Environ 31: 3–10.
Simpson B . (1979). Quaternary biogeography of the high montane regions of South America. In: Duellman WE (ed). The South American Herpetofauna: its Origin, Evolution, and Dispersal. Monograph of the Museum of Natural History, University of Kansas: Lawrence, Kansas. pp 157–188.
Solà E, Sluys R, Gritzalis K, Riutort M . (2013). Fluvial basin history in the northeastern Mediterranean region underlies dispersal and speciation patterns in the genus Dugesia (Platyhelminthes, Tricladida, Dugesiidae). Mol Phyl Evol 66: 877–888.
Suguio K, Martin L . (1978). Quaternary marine formations of the state of São Paulo and southern Rio de Janeiro. Int Symp Coastal Evol Quat 1: 1–55.
Sunnucks P, Blacket MJ, Taylor JM, Sands CJ, Ciavaglia SA, Garrick RC et al. (2006). A tale of two flatties: different responses of two terrestrial flatworms to past environmental climatic fluctuations at Tallaganda in montane southeastern Australia. Mol Ecol 15: 4513–4531.
Tajima F . (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595.
Thomé MTC, Zamudio KR, Giovanelli JGR, Haddad CFB, Baldissera FA, Alexandrino J . (2010). Phylogeography of endemic toads and post-Pliocene persistence of the Brazilian Atlantic Forest. Mol Phyl Evol 55: 1018–1031.
Wegmann D, Leuenberger C, Neuenschwander S, Excoffier L . (2010). ABCtoolbox: a versatile toolkit for approximate Bayesian computations. BMC Bioinformatics 11: 116.
Winsor L, Johns PM, Barker GM . (2004). Terrestrial planarians (Platyhelminthes: Tricladida: Terricola) predaceous on terrestrial gastropods. In: Barker GM (ed). Natural Enemies of Terrestrial Molluscs. CABI Publishing: Oxfordshire, UK. pp 227–278.
Acknowledgements
This research was supported by Fundación BBVA of Spain, the Ministerio de Ciencia e Innovación of Spain and FAPESP (Brazil). ASG was supported by a grant under the program Beatriu de Pinós (2010BP-B 00175). We thank Instituto Chico Mendes de Conservação da Biodiversidade (ICMBio), Instituto Estadual do Ambiente do Governo do Rio de Janeiro (INEA), Instituto Florestal do Governo do Estado de São Paulo (IF), Museu de Zoologia da Universidade de São Paulo (MZUSP), Fundação do Meio Ambiente do Governo Estado de Santa Catarina (FATMA) for sampling licences in Reserva Biológica Augusto Ruschi, Parque Nacional da Serra dos Órgãos, Parque Nacional Serra da Bocaina, Parque Nacional Saint Hilaire/Lange, Parque Nacional da Serra de Itajaí, Floresta Nacional de São Francisco de Paula (ICMBio); Parque Estadual do Desengano (INEA); Parque Estadual de Campos do Jordão, Parque Estadual Serra da Cantareira, Parque Estadual Intervales (IF), Estação Biológica de Boraceia (MZUSP); Parque Estadual da Serra do Tabuleiro (FATMA). We thank Alceli Rodrigues, Júlio Pedroni, Débora Redivo, Cláudia Olivares, José Grau, Marília Jucá, Leonardo Zerbone and Welton Araújo for their help with sampling, Ana Cristina Machado Vasconcelos, Italo D’Elia, Ana Cavalcanti and Lígia Domingos for the histological sectioning and staining, Sebastian Ramos-Onsins for assistance with the mlcoalsim program, Joan Garcia-Porta and Leticia Bidegaray for support in niche modelling analyses, Daniel Wegmann for helpful advice with the ABC-GLM method, A Carnaval and C Moritz for kindly providing us with the details of their paleoclimate models and Gema Blasco for technical support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Rights and permissions
About this article
Cite this article
Álvarez-Presas, M., Sánchez-Gracia, A., Carbayo, F. et al. Insights into the origin and distribution of biodiversity in the Brazilian Atlantic forest hot spot: a statistical phylogeographic study using a low-dispersal organism. Heredity 112, 656–665 (2014). https://doi.org/10.1038/hdy.2014.3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2014.3
Keywords
This article is cited by
-
Diet assessment of two land planarian species using high-throughput sequencing data
Scientific Reports (2019)
-
Phylogeography of ants from the Brazilian Atlantic Forest
Organisms Diversity & Evolution (2019)
-
Cryptic diversity in an Atlantic Forest malaria vector from the mountains of South-East Brazil
Parasites & Vectors (2018)
-
Rolling into the deep of the land planarian genus Choeradoplana (Tricladida, Continenticola, Geoplanidae) taxonomy
Organisms Diversity & Evolution (2018)
-
Ancient divergence and recent population expansion in a leaf frog endemic to the southern Brazilian Atlantic forest
Organisms Diversity & Evolution (2015)
