
Abstract
Elucidating the demographic and landscape features that determine the genetic effects of habitat fragmentation has become fundamental to research in conservation and evolutionary biology. Land-bridge islands provide ideal study areas for investigating the genetic effects of habitat fragmentation at different temporal and spatial scales. In this context, we compared patterns of nuclear microsatellite variation between insular populations of a shrub of evergreen broad-leaved forest, Loropetalum chinense, from the artificially created Thousand-Island Lake (TIL) and the Holocene-dated Zhoushan Archipelago of Southeast China. Populations from the TIL region harboured higher levels of genetic diversity than those from the Zhoushan Archipelago, but these differences were not significant. There was no correlation between genetic diversity and most island features, excepting a negative effect of mainland–island distance on allelic richness and expected heterozygosity in the Zhoushan Archipelago. In general, levels of gene flow among island populations were moderate to high, and tests of alternative models of population history strongly favoured a gene flow-drift model over a pure drift model in each region. In sum, our results showed no obvious genetic effects of habitat fragmentation due to recent (artificial) or past (natural) island formation. Rather, they highlight the importance of gene flow (most likely via seed) in maintaining genetic variation and preventing inter-population differentiation in the face of habitat ‘insularization’ at different temporal and spatial scales.
Similar content being viewed by others

Introduction
Understanding the effects of fragmentation and associated demographic changes on genetic variation and also population viability has become increasingly important to conservation biologists and population geneticists (Cornuet and Luikart, 1996; Luikart and Cornuet, 1998). A lack of connectivity, particularly in small and isolated populations, leads to a multitude of demographic and genetic consequences, including a loss of genetic variation (Lande, 1988), increased inbreeding, increased susceptibility to stochastic processes and increased likelihood of extinction (Frankel and Soulé, 1981; Lande, 1994; Garza and Williamson, 2001). As a general guideline, genetic variability decreases at a constant rate of 1/(2Ne) per generation in a closed population (where Ne is the effective population size; Wright, 1978). Thus, loss of genetic diversity is predicted to occur much more rapidly in smaller populations because of their sensitivity to demographic and environmental stochasticity and drift (Amos and Balmford, 2001). However, levels of genetic variability can also recover due to rapid post-bottleneck population growth (Nei et al., 1975) or even small amounts of immigration (Keller et al., 2001). Moreover, levels of genetic diversity in post-fragmentation populations are also highly influenced by life-history traits, in particular longevity and mating system (Kuo and Janzen, 2004; Hailer et al., 2006) and characteristics of the landscape (Wang et al., 2014). For example, long generation times slow the effective rate of genetic drift if habitat fragmentation has occurred only recently (for example, <200 years ago), thus providing an ‘intrinsic buffer’ to the loss of genetic diversity (Hailer et al., 2006). These and other factors indicate that simple deterministic equations or generalized principles are of limited use for forecasting the loss in genetic variation over more than a single generation bottleneck (England et al., 2003). Instead, our understanding of the process is perhaps best pursued through case-specific studies at different temporal and spatial scales.
In this regard, the clear boundaries and the effective isolation of water, as well as isolation at different time periods make land-bridge island systems ideal candidates for studies that examine fundamental processes associated with the maintenance of genetic variation (White and Searle, 2007). Land-bridge islands may be classified as either continental shelf islands or artificial-lake islands. The former were originally part of a nearby continent but were separated by rising sea levels, for example, during post-glacial times (Whittaker and Fernández-Palacios, 2007). By contrast, artificial-lake islands are often formed by dam construction over very recent time scales. These islands represent former hilltops that were not flooded by the lake. Although there are numerous studies of genetic diversity on land-bridge islands in America (Jordan and Snell, 2008; Barker et al., 2012; Bell et al., 2012), Australia (Furlan et al., 2012; Stankowski and Johnson, 2014) and the Mediterranean Basin (Bittkau and Comes, 2005), relatively little attention has been paid to China (Liu et al., 2013; Wang et al., 2014), despite the fact that China has >10 000 islands, with climates ranging from tropical to cold temperate conditions (Committee ZGHDZ, 2013).
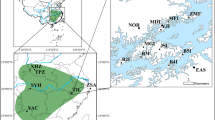
The Thousand-Island Lake (TIL), located in Southeast China (Chunan County, Zhejiang Province), is a large (ca 573 km2) artificial reservoir, which was formed in 1959 by damming of the Xinanjiang River for constructing a hydroelectric power station (see Figure 1). After the dam closure, the water level raised fast, flooded former mountains and created >1000 islands of different sizes (0.25–1320 ha), covering about 83 km2 in total (Wang et al., 2009). By contrast, the Zhoushan Archipelago, located in the East China Sea to the south of the Yangtze River Delta (Figure 1), was originally an extended part of the Tiantai Mountains (eastern Zhejiang Province), and separated from the mainland by rising sea levels about 7000–9000 years ago (Wang and Wang, 1980; Zhou, 1987). It is the largest (ca 20 800 km2) archipelago of China, consisting of 1339 islands and 3306 reefs, which cover about 1440 km2 in total (Zhou, 1987). The vegetation of the TIL and Zhoushan islands mainly comprises subtropical evergreen broad-leaved forest, with Fagaceae, Hamamelidaceae and Theaceae dominating (Luo et al., 2012). However, these two island systems vary greatly in spatial scale and temporal origin, which makes them ideal systems for a comparative study of disentangling the effects of geography (for example, island area, mainland–island distance) and history (time since island isolation) on levels of genetic variation (see also Capula and Ceccarelli, 2003).

Sample localities of L. chinense in the TIL region and the Zhoushan Archipelago of Southeast China. Population codes are identified in Table 1.
Loropetalum chinense (R.Br.) Oliv. (Hamamelidaceae) is a diploid (2n=24), evergreen shrub or small tree and a major component of the subtropical evergreen broad-leaved forest of the lower mountains (ca 300–600 m above sea level) of Southeast China (Lian and Xiao, 2001; Zhang et al., 2003). Individuals typically require 5–8 years to reach the reproductive stage, and genets may persist for decades (YX Qiu, personal observation). The pollination biology of L. chinense has not been reported yet. However, the bisexual flowers of this species are possibly pollinated by flies, as reported for the closely related L. subcordatum (Benth.) Oliv. (Gu and Zhang, 2008). The fruit of L. chinense is a dehiscing, two-seeded capsule. According to Zhang et al. (2003), this species exhibits ballistic seed dispersal (similar to Hamamelis spp.). However, there is insufficient knowledge on the pollen and seed dispersal ability of L. chinense, which makes it difficult to predict the effects of artificial and natural habitat fragmentation on the species’ gene flow patterns and genetic structure in the TIL region and the Zhoushan Archipelago, respectively.
Here, we use nuclear microsatellite (nSSR) markers to examine patterns of genetic diversity and differentiation across six island populations of L. chinense each in the TIL region and the Zhoushan Archipelago, respectively. Specifically, our goals were (i) to detect the effects of artificial versus natural habitat fragmentation on the species’ gene flow patterns and genetic structure in the two island systems; (ii) to evaluate the relative importance of island area, island population size, time since island isolation and distance to the mainland on the genetic diversity of the island populations; and (iii) to examine potential effects of demographic history versus gene dispersal characteristics on the genetic diversity of L. chinense. This comparative study of two island systems with different landscape features and histories will increase our understanding of evolutionary and population dynamic processes in fragmented habitats.
Materials and methods
Sample localities and microsatellite genotyping
Leaf material was collected in 12 L. chinense populations (381 individuals), representing six islands each in the TIL region and the Zhoushan Archipelago, respectively (Table 1; Figure 1). At least 21 individuals per population were sampled except for population ZS5, which was represented by seven individuals. Total genomic DNA was extracted from the silica gel-dried leaf material using DNA Plantzol (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. Variation was assayed at eight microsatellite loci (Lc01, Lc02, Lc04, Lc11, Lc18, Lc25, Lc31 and Lc38) specifically developed for L. chinense (GenBank accession numbers: KJ830134–KJ830127). Primers were labeled with a fluorescent dye, either HEX or 6-FAM (Applied Biosystems, Foster City, CA, USA), and PCR amplifications were performed on a GeneAmp 9700 DNA Thermal Cycler (Perkin-Elmer, Waltham, MA, USA) using a 10 μl reaction mixture containing 30–50 ng μl–1 of genomic DNA, 0.5 U of Taq polymerase (TaKaRa Biotechnology Co., Ltd, Dalian, China), 1 μl of 10 × PCR buffer with MgCl2, 1 μl of dNTPs (2.5 mM each) and 0.5 μl of each primer (10 μM). PCR amplification conditions were as follows: initial denaturation at 95 °C for 5 min; 30 cycles of 30 s at 95 °C, 30 s at the optimized annealing temperature, 45 s of elongation at 72 °C; ending with a 10-min extension at 72 °C. PCR products were separated on a MegaBACE 1000 (GE Healthcare Biosciences, Pittsburgh, PA, USA). Alleles were scored manually with the aid of the software GENETIC PROFILER (version 2.2; GE Healthcare Biosciences).
Genetic diversity analyses of the sampled loci and populations
We used FSTAT (version 2.9.3; Goudet, 1995), which estimates genetic measures that are corrected for different sample size, to calculate the following parameters for each locus: the observed number of alleles (A), observed heterozygosity within populations (HO), expected heterozygosity within populations (HS) under Hardy–Weinberg equilibrium (HWE), and total genetic diversity over populations (HT). Within each population, linkage disequilibrium was tested between loci using exact tests with 10 000 dememorizations, 1000 batches and 10 000 iterations performed with GENEPOP (http://www.genepop.curtin.edu.au/). Statistical significance (α=0.05) was evaluated based on 1000 permutations, and then corrected for multiple tests using the sequential Bonferroni method (Rice, 1989). Null allele frequencies were obtained using the program MICRO-CHECKER (version 2.2.3; Van Oosterhout et al., 2004). For each population, the following genetic diversity parameters were estimated across all loci: the mean number of alleles (NA), allelic richness (AR, standardized for the minimum sample size of seven individuals using rarefaction; Petit et al., 1998), HO and expected heterozygosity (HE) under HWE. Departures from HWE, given by the deviation of within-population inbreeding coefficients (FIS) from zero, were tested by 10 000 permutations. Differences in parameter estimates between populations from the TIL region and Zhoushan Archipelago were tested using a 10 000 permutation procedure implemented in FSTAT, with populations allocated at random to these alternative regions.
Analyses of population genetic structure and isolation-by-distance
Genetic structure was quantified by among-population FST (Weir and Cockerham, 1984) using FSTAT. The significance of FST (between pairs of populations and overall) was assessed by 1000 random permutations across loci, followed by Bonferroni correction for pairwise results. Hierarchical analysis of molecular variance in ARLEQUIN (version 3.1; Excoffier et al., 2005) was used to quantify the partitioning of genetic variance at three levels (that is, among regions, among populations within regions and within populations). The significance of each variance component was tested through 10 000 permutations.
We used the Bayesian clustering analysis implemented in STRUCTURE (version 2.3.1; Pritchard et al., 2000) to infer the number of distinct genetic groups observed in our data. The analyses were run using the admixture model with independent allele frequencies. The number of K was set to vary from 1 to 12. For each value of K, we used a burn-in of 10 000 and a Markov chain Monte Carlo length of 100 000 iterations. The upper limit of 12 was chosen as this corresponds to the number of sampled populations. Ten independent simulations were performed of each K to check for consistency across runs. The most likely number of clusters was chosen by calculating ΔK (Evanno et al., 2005), which is the second-order rate of change of the mean loglikelihood of the data [LnP(D)] between successive K values in STRUCTURE HARVESTER (version 0.6.8; Earl and VonHoldt, 2012). When K was defined, the run with the highest LnP(D) value was selected and individuals were assigned to clusters based on maximum membership proportions (Q>0.80).
We evaluated the relative influences of migration and drift on population structure in each region by correlating FST/(1–FST) with geographical distance and testing for statistical significance with 1000 permutations of Mantel test in IBDW (version 3.23; Jensen et al., 2005). A significant linear relationship of increased genetic differentiation at greater geographical distances (isolation-by-distance) is expected under the stepping-stone model of gene flow when the opposing forces of migration and drift are in equilibrium (Johnson et al., 2003; Jordan and Snell, 2008). In contrast, a nonsignificant linear relationship between genetic differentiation and geographical distance combined with large variance in pairwise FST is expected for a scenario of drift under extreme population isolation (Hutchison and Templeton, 1999).
Tests of relationships between island features and genetic diversity
To better understand which island features contribute to genetic diversity, we used linear regressions to test for each region whether diversity (that is, NA, AR, HO and HE) was positively correlated with island area but negatively with the geographical distance of islands to the nearest mainland. For the Zhoushan Archipelago, where (in contrast to the TIL region) islands became isolated at different times, we also tested for a negative correlation between genetic diversity and time since island isolation. The islands sampled ranged in area from 0.25 (ha) to 1320 (ha) in the TIL region and from 694 (ha) to 50 265 (ha) in the Zhoushan Archipelago (Table 1). Distances of each island to the mainland (see Table 1) were measured as minimum distances using Google Earth (http://earth.google.com). The time since island isolation was estimated based on minimum ocean depths (the depth of the underwater saddles that connect adjacent islands or the mainland) and data on the rates of sea level rise since the last glacial maximum (ca <21 000 years ago; Foufopoulos and Ives, 1999). Based on the minimum isobaths values of these islands (NGDCNH, 2012) and the change curve of the East China Sea level since the late Pleistocene (Guo, 1979), we calculated the time since isolation of each island in the Zhoushan Archipelago. The time since island isolation was estimated to range from 8150 years before present for ZS2 to 8750 years before present for ZS4 (Table 1).
Estimation of contemporary gene flow
We estimated contemporary gene flow among insular populations in each region using the Bayesian method implemented in BAYESASS (version 1.3; Wilson and Rannala, 2003). In a first step, we adjusted the delta values for allele frequencies, migration rates and inbreeding coefficients to ensure that acceptance rates for changes in these parameters fell between 40 and 60% (Wilson and Rannala, 2003). We then ran the program for 1 × 107 iterations, including a burn-in of 106 generations. Model convergence was assessed by comparison of posterior probability densities of inbreeding coefficients and allele frequencies across 10 replicate runs, each with a different initial seed. As recommended in a recent evaluation of BAYESASS (Faubet et al., 2007), the Bayesian deviance measure was used to determine the run that displayed the best model fit (Spiegelhalter, 2002). For this best-fit run, we then ran the analysis again using the seed from the best-fit run but increased the run length to 5 × 107 iterations. The results presented were from this final run.
Demographic analyses
We used two methods implemented in BOTTLENECK (version 1.2.02; Piry et al., 1999) to detect genetic bottlenecks due to the different time scales associated with each method (Cornuet and Luikart, 1996; Garza and Williamson, 2001; Spear et al., 2006). First, we used Wilcoxon’s sign-rank test, which examines whether populations exhibit a greater level of heterozygosity than predicted in a population at mutation-drift equilibrium. This test is most sensitive at detecting bottlenecks occurring over approximately the last 2–4 Ne generations. Second, we used the mode-shift test, which is most appropriate for detecting more recent population declines, specifically over the last few dozen generations (Cornuet and Luikart, 1996; Luikart et al., 1998). This test assumes that non-bottlenecked populations at mutation-drift equilibrium have a large proportion of alleles at low frequency and a smaller proportion of alleles at intermediate frequencies (L-shaped distribution) (Luikart et al., 1998). In bottlenecked populations, the distribution will shift toward one in which a smaller proportion of alleles are found at low frequency (<10%) than at intermediate frequency because the alleles at low frequency are those most likely lost during a bottleneck (shifted mode distribution). For each population, we performed 10 000 simulations under both the stepwise mutation model and the two-phase model with 95% single-step mutations and 5% multi-step mutations, as recommended by Piry et al. (1999). P-values from the Wilcoxon test were used as evidence for bottlenecks occurring at each timescale and were assessed for significance at the 0.05 level.
Finally, we evaluated which of two models of population history (gene flow versus drift only) best describes the processes leading to the current population structure of the two regions. For this purpose, we used the software 2MOD (version 0.2; Ciofi et al., 1999), which estimates the relative likelihoods of each model using coalescent-based Markov chain Monte Carlo simulations. The gene flow model assumes populations are at drift-migration equilibrium and uses gene frequencies to estimate the relative strength of drift versus gene flow for each population. By contrast, the drift model assumes a historically panmictic population separated into many smaller populations that have since been evolving independently through drift alone in the absence of gene flow. Both models assume that mutation is a negligible factor and calculate a parameter F, which indicates the probability of allelic co-ancestry in a given population. We ran the program twice for each model with 100 000 Markov chain Monte Carlo updates; the first 10% of the output was discarded as burn-in. Results from the two runs were combined and the probability of each model was calculated as the number of draws for a given model out of the total draws. We used Bayes factors to describe the probability of the most likely model over the probability of the other model.
Results
Characteristics of the nSSR loci
Estimates of diversity varied among the eight nSSR loci surveyed (Table 2). The number of alleles per locus (A) varied between 5 and 25, with a total of 126 alleles detected overall. The average value of expected within-population diversity (HS) among loci was 0.62, with a range of 0.13–0.90. Finally, per locus estimates of total genetic diversity over all populations (HT) ranged from 0.14 to 0.92 and averaged 0.65 (Table 2). MICRO-CHECKER analysis provided no evidence of scoring errors due to large allele drop-out or stutter peaks in our final data set. However, a low frequency of null alleles (⩽0.11) was detected in four populations (TL1, TL4, ZS2 and ZS3). There was no evidence of linkage disequilibrium between any pair of loci across populations. Of 96 population-by-locus tests, 27 (28.1%) deviated significantly from HWE after sequential Bonferonni correction. However, these were not limited to a single locus or sampling site and so all eight loci were retained for further analysis. Heterozygote deficiency was detected in five populations (TL1, TL4, ZS1, ZS4 and ZS6) at one to five loci, resulting in statistically positive FIS values, although low (⩽0.102) (see Table 1).
Analyses of population genetic diversity and structure
Within populations, the mean number of alleles (NA) ranged from 8.3 to 9.4 in the TIL region and from 4.9 to 9.9 in the Zhoushan Archipelago (Table 1). Mean allelic richness (AR) was 5.40 in the former and 5.16 in the latter region. Values of observed (HO) and expected heterozygosity (HE) were very similar, with HE ranging between 0.57 and 0.70, and averaging 0.64 and 0.59 for the TIL region and the Zhoushan Archipelago, respectively (Table 1). The six populations from the TIL region had higher levels of diversity (mean: AR=5.398; HO=0.629; HE=0.652) than those from the Zhoushan Archipelago (mean: AR=5.163; HO=0.563; HE=0.597), but these differences were not significant (P=0.050–0.098) (Table 3). Levels of inbreeding in both regions were close to zero (0.035 vs 0.056) and not significantly different from each other (P=0.445) (Table 3).
Pairwise estimates of FST ranged from 0.005 to 0.175 (Supplementary Appendix S1), with an overall value of 0.048, indicating low levels of genetic differentiation over all populations. Of the 66 pairwise comparisons, 60 were significant after sequential Bonferroni correction (Supplementary Appendix S1). Mean pairwise FST was 0.061 in the TIL region and 0.021 in the Zhoushan Archipelago, but the difference between the two regions was not significant (P=0.459). In each region, there was no pattern of isolation-by-distance (TIL: r=−0.416, P=0.865; Zhoushan Archipelago: r=0.463, P=0.898).
Based on the STRUCTURE analysis, the most appropriate K for L. chinense was three (Supplementary Appendix S2). Figure 2 shows the assignment of individuals to clusters I (‘red’), II (‘green’) and III (‘blue’), and the admixture proportions of each individual. Clusters I and II had a high vs moderate frequency among individuals from the TIL region (41% vs 26% of all local samples), whereas the majority of individuals in the Zhoushan Archipelago were more or less equally assigned to clusters II and III (32.6% vs 45.5%). Notably, in the TIL region, a geographically isolated population, TL5 (Figure 1), had the great majority of individuals (94%) assigned to cluster I, which may reflect regional gene pool subdivision predating island formation (see also Yuan et al., 2012). Hierarchical analysis of molecular variance attributed only a low percentage of the total genetic variance (0.94%) to differences among the two island regions, compared with 4.38% among populations within these regions, and 94.68% within populations (Table 4).

Histogram of the STRUCTURE analysis for the model with K=3 (showing the highest ΔK) based on the nSSR data of 381 individuals (12 populations) of L. chinense from the TIL region and the Zhoushan Archipelago. Each colour corresponds to a suggested cluster, and a vertical bar represents a single individual. The x axis corresponds to population codes. The y axis presents the estimated membership coefficient (Q) for each individual in the three clusters.
Effects of island features on genetic diversity
In the TIL region, neither island area nor distance to the mainland were correlated with genetic diversity (that is, NA, AR, HO, HE). In the Zhoushan Archipelago, mainland–island distance was negatively correlated with both allelic richness (AR) (r=−0.854, P=0.015) and expected heterozygosity (HE) (r=−0.732, P=0.049) (Supplementary Appendix S3), whereas none of the genetic diversity parameters were related to island area or time since island isolation.
Contemporary inter-island gene flow
BAYESASS uses a Bayesian approach and Markov chain Monte Carlo sampling to generate mc values, which reflect migration rates over ‘the last few generations’ (Wilson and Rannala, 2003). Our Bayesian estimates of contemporary migration rates (mc) varied from 0.009 to 0.187 in the TIL region and from 0.006 to 0.162 in the Zhoushan Archipelago (Supplementary Appendix S4). In the latter region, population ZS1 from the largest island (‘Zhoushan’) served as a main source of gene flow to other insular populations (mc=0.089–0.162). Except for one population, TL1, which was largely composed of individuals (94%) that originated from within the same site, we detected moderate to high inter-population migration rates (that is, gene flow rates) with ca 20–30% of the individuals in the majority of populations being exchanged with other sites in both regions (Supplementary Appendix S4).
According to the 2MOD analysis, the gene flow-drift model was much more strongly favoured than a pure drift model at the species level (P (gene flow model)=1.0, Bayes factor=100 000). When the two regions were considered separately, there was still no role for a drift-only model (P (gene flow model)=1.0 in each case).
Changes in population size
The Wilcoxon test as implemented in BOTTLENECK under the stepwise mutation model and two-phase model model did not detect any recent genetic bottleneck in either region, and all populations showed L-shaped allelic distributions, as expected in the absence of a recent bottleneck (Table 5).
Discussion
Potential effects on the genetic diversity of L. chinense: demographic history vs gene dispersal characteristics
In this study, we compared the population genetic diversity and structure of L. chinense between two contrasting island systems. Our results showed that the mean diversity values of populations from the Zhoushan Archipelago were lower than the estimates of populations from the TIL region, but the differences were not significant (P=0.050–0.098) (Table 3). It remains unknown how much of the initial genetic variation has been lost in each region following island formation; however, by comparison with the TIL region (viz a 55-year time period of habitat fragmentation), these results may suggest that populations in the Zhoushan Archipelago have retained a considerable proportion of their initial (pre-fragmentation) genetic diversity over extended time periods (that is, thousands of years) and to the present day (see below).
In general, the genetic effects of habitat fragmentation on plant species vary depending on their life-history traits. For example, for short-lived herbs, genetic erosion may occur over very short time scales, while for tree species, the long generation time may buffer the negative genetic effects of fragmentation for hundred or even thousand years (for example, Aquilar et al., 2008). Hence, it is often difficult to make predictions about the genetic effects of habitat fragmentation and ascertain the time threshold for negative demographic effects to become apparent (Young et al., 1996; Kramer et al., 2008). A previously reported negative impact of fragmentation on genetic diversity was found in Hedyotis chrysotricha (Rubiaceae), a short-lived (probably annual) herb, in the TIL region (Yuan et al., 2012). By contrast, our inference of no significant effect of habitat fragmentation on the genetic diversity of L. chinense, even over the last thousands of years, may be explained by demographic factors and/or species-specific gene dispersal characteristics.
In terms of demographics, we detected deviations from HWE in 5 of the 12 populations studied (Table 1). However, overall levels of inbreeding in both island regions were close to zero (Table 3) and none of the populations showed genetic signatures of recent bottlenecks (Table 5). In addition, the 2MOD analysis clearly favoured a gene flow-drift model over a pure drift model in either region and at the species level. It is therefore likely that population sizes of L. chinense have remained sufficiently large as island formation that prevents loss of genetic diversity via inbreeding and genetic drift (for example, Harrison and Hastings, 1996; Schaal and Leverich, 1996; Ewers and Didham, 2006). For the TIL region, of course, a mutually non-exclusive explanation is that insufficient time since island formation has elapsed for such effects to become measurable. In any event, the present results provide little evidence that demographic factors had a major role in the post-fragmentation history of L. chinense.
In terms of gene dispersal, one also has to consider that the ability of pollen and seeds to move through a fragmented landscape will determine the potential of a plant species to counteract the effects of genetic drift (Young et al., 1996). However, due to the limited pollen-dispersal capabilities of flies, which likely dominate the pollinator spectrum of L. chinense (Gu and Zhang, 2008; see Introduction section), it seems unlikely that gene dispersal through pollen alone has been sufficient to promote significant levels of gene flow and maintain the species’ population connectivity across the islands. By contrast, the small (ca 4–5 mm) seeds of L. chinense are not only dispersed ballistically and by wind but possibly also through floating water, as the seed coat is hard and impermeable to water and gas (Zhang et al., 2003). However, in the absence of detailed morphological and experimental evidence, hydrochory in L. chinense must remain speculative at present. This caveat notwithstanding, and as further discussed below, the genetic evidence presented here suggests that this species has a high seed dispersal capability to maintain moderate to high levels of ongoing gene flow.
Similar genetic structures and gene flow patterns across island systems
Despite the highly fragmented landscape occupied by L. chinense, we found little evidence of strong genetic structuring in this evergreen broad-leaved forest species within and across the two island systems. First, the great majority of populations could not be confidently assigned to any single cluster in the Bayesian clustering analysis (Figure 2). Second, the overall FST value calculated (0.048) indicated low levels of genetic differentiation between populations (Supplementary Appendix S1). And finally, the largest proportion of genetic variance resided within populations (94.68%), whereas the among-region component was low (0.94%) and nonsignificant (P=0.054; Table 4). When further combined with the lack of isolation-by-distance in each island system, these results already suggest that L. chinense is a high gene flow species with non-equilibrium population structures in both island systems, regardless of their significant differences in spatial-temporal characteristics.
As to more precise estimates, our BAYESASS analyses indeed showed that contemporary migration rates among recently fragmented populations in the TIL region were moderate to high (mc=0.009–0.187), whereby individuals of most populations had ca 20% probability of being exchanged with other sites (Supplementary Appendix S4). However, given the much longer time since island isolation in the Zhoushan Archipelago (about 7000–9000 years ago), and the larger geographic distance between sampled populations there (Figure 1), one may have expected higher population differentiation (and correspondingly lower inter-island gene flow), but this was not observed. Instead, as the most conspicuous result of this study, populations in the Zhoushan Archipelago exhibited low levels of differentiation like their conspecifics in the TIL region (FST=0.021 vs 0.061). Similarly, moderate to high levels of gene flow among insular populations (mc=0.006–0.162) were observed in the Zhoushan Archipelago (individuals of most populations had ca 30% probability of being exchanged with other sites, Supplementary Appendix S4). Interestingly, population ZS1 from the largest island of this archipelago (‘Zhoushan’) provided the main source of gene flow to other insular populations. Overall, these results support the notion that L. chinense has a high potential for gene flow (most likely via seed dispersal), and that patterns of gene flow and population connectivity in this species have not been greatly modified by either recent (artificial) or past (natural) fragmentation of their forested habitats.
No major effects of island characteristics on genetic diversity
In contrast to recent studies on animal systems (fig wasps: Liu et al., 2013; pond frogs: Wang et al., 2014), we did not find a significant relation between genetic diversity and time since island isolation in the Zhoushan Archipelago, and the same was true for island area in both regions. Nonetheless, we found negative correlations between mainland–island distance and both allelic richness (AR) and expected heterozygosity (HE) in the Zhoushan Archipelago but not in the TIL region, which may simply reflect differences in spatial scale. However, given the larger island areas of the Zhoushan Archipelago, it is still puzzling why populations of L. chinense there harbour similar, rather than higher levels of genetic diversity compared with those from the TIL region. Perhaps the most likely explanation is that the archipelago populations have experienced genetic erosion through recent bottleneck, but which have gone undetected because of lack of statistical power (particularly as we used a limited number of nSSR loci; see also Yuan et al., 2012). This hypothesis is further suggested by the fact that the natural landscapes of the Zhoushan Archipelago have been greatly modified by human activities over the last 20 years, whereby the impervious surface area continues to increase at an average rate of 1.97 km2 per year (Zhang et al., 2013). Although certain agricultural developments also may have positive effects for some organisms, for example, frogs, for which irrigated fields provide suitable habitats (Knutson et al., 2004; Wang et al., 2014), much of the natural vegetation cover (that is, subtropical evergreen broad-leaved forest) has now been destroyed (Zhang et al., 2013). Evidently, more fine-scale studies, using precise deforestation records, highly resolving molecular (for example, genomics) approaches, and a dense sampling, are required to determine whether such recent forest fragmentation has already affected the genetic architecture of constituent species, such as L. chinense.
Conclusions
The genetic consequences of habitat fragmentation remain a critical issue in both conservation and evolutionary biology. Our comparative study of L. chinense populations in the TIL region and the Zhoushan Archipelago shows no obvious genetic effects of forest fragmentation as a result of recent (artificial) and past (natural) island formation, respectively. These findings contrast with recent studies in either region, indicating that recent artificial ‘islanding’ in the TIL region can lead to significant loss of genetic diversity, especially in short-lived herbs (Yuan et al., 2012). Moreover, in the Zhoushan Archipelago, it was recently demonstrated that overwater distance is a significant barrier to contemporary gene flow in both pond frogs (Wang et al., 2014) and fig wasps (Liu et al., 2013), whereby in either instance genetic drift following Holocene island formation likely decreased diversity and increased differentiation among insular populations. By contrast, this study on L. chinense is the first to highlight the importance of gene flow (most likely via seed) in sustaining genetic diversity and preventing inter-population differentiation in the face of habitat ‘insularization’ in both the TIL region and the Zhoushan Archipelago, that is, at different temporal and spatial scales. In this context, both island systems should figure prominently in future (for example, coalescent-based) studies addressing the genetic consequences of subtropical forest fragmentation, with the challenge of precisely estimating pre-fragmentation parameters (for example, Ne) for comparison with present conditions (Srikwan and Woodruff, 2000).
Data archiving
Data have been deposited in Dryad (http://doi.org/10.5061/dryad.585t1).
Accession codes
References
Aguilar R, Quesada M, Ashworth L, Herrerias-Diego Y, Lobo J . (2008). Genetic consequences of habitat fragmentation in plant populations: susceptible signals in plant traits and methodological approaches. Mol Ecol 17: 5177–5188.
Amos W, Balmford A . (2001). When does conservation genetics matter? Heredity 87: 257–265.
Barker BS, Rodriguez-Robles JA, Aran VS, Montoya A, Waide RB, Cook JA . (2012). Sea level, topography and island diversity: phylogeography of the Puerto Rican Red–eyed Coqui, Eleutherodactylus antillensis. Mol Ecol 21: 6033–6052.
Bell RC, Brasileiro CA, Haddad CFB, Zamudio KR . (2012). Evolutionary history of Scinax treefrogs on land-bridge islands in south-eastern Brazil. J Biogeogr 39: 1733–1742.
Bittkau C, Comes HP . (2005). Evolutionary processes in a continental island system: molecular phylogeography of the Aegean Nigella arvensis alliance (Ranunculaceae) inferred from chloroplast DNA. Mol Ecol 14: 4065–4083.
Capula M, Ceccarelli A . (2003). Distribution on genetic variation and taxonomy of insular and mainland populations of the Italian wall lizard, Podarcis sicula. Amphibia–Reptilia 24: 483–495.
Ciofi C, Beaumont MA, Singland IR, Bruford MW . (1999). Genetic divergence and units for conservation in the Komodo dragon Varanus komodoensis. P Roy Soc Lond B Bio 266: 2269–2274.
Committee ZGHDZ. (2013) Zhong Guo Hai Dao Zhi. China Ocean Press: Beijing, China.
Cornuet JM, Luikart G . (1996). Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144: 2001–2014.
Earl DA, vonHoldt BM . (2012). Structure harvester: a website and program for visualizing structure output and implementing the Evanno method. Conserv Genet Resour 4: 359–361.
England PR, Osler GH, Woodworth LM, Montgomery ME, Briscoe DA, Frankham R . (2003). Effects of intense versus diffuse population bottlenecks on microsatellite genetic diversity and evolutionary potential. Conserv Genet 4: 595–604.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Laval G, Schneider S . (2005). Arlequin version 3.0: an integrated software package for population genetics data analysis. Evolutionary bioinformatics online 1: 47–50.
Ewers RM, Didham RK . (2006). Confounding factors in the detection of species responses to habitat fragmentation. Biol Rev 81: 117–142.
Faubet P, Waples RS, Gaggiotti OE . (2007). Evaluating the performance of a multilocus Bayesian method for the estimation of migration rates. Mol Ecol 16: 1149–1166.
Foufopoulos J, Ives AR . (1999). Reptile extinctions on land-bridge islands: life-history attributes and vulnerability to extinction. Am Nat 153: 1–25.
Frankel OH, Soulé ME . (1981) Conservation and Evolution. Cambridge University Press: Cambridge, UK.
Furlan E, Stoklosa J, Griffiths J, Gust N, Ellis R, Huggins RM et al. (2012). Small population size and extremely low levels of genetic diversity in island populations of the platypus, Ornithorhynchus anatinus. Ecol Evol 2: 844–857.
Garza JC, Williamson EG . (2001). Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10: 305–318.
Goudet J . (1995). FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered 86: 485–486.
Gu L, Zhang DX . (2008). Autogamy of an endangered species: Loropetalum subcordatum (Hamamelidaceae). J Syst Evol 46: 651–657.
Guo XD . (1979). Sea level changes since late Pleistocene in China. Chin J Geol 4: 330–341.
Hailer F, Helander B, Folkestad AO, Ganusevich SA, Garstad S, Hauff P et al. (2006). Bottlenecked but long-lived: high genetic diversity retained in white-tailed eagles upon recovery from population decline. Biol Lett 2: 316–319.
Harrison S, Hastings A . (1996). Genetic and evolutionary consequences of metapopulation structure. Trends Ecol Evol 11: 180–183.
Hutchison DW, Templeton AR . (1999). Correlation of pairwise genetic and geographic distance measures: inferring the relative influence of gene flow and drift on the distribution of genetic variability. Evolution 53: 1898–1914.
Jensen JL, Bohonak AJ, Kelley ST . (2005). Isolation by distance, web service. BMC Genet 6: 13.
Johnson JA, Toepfer JE, Dunn P . (2003). Contrasting patterns of mitochondrial and microsatellite population structure in fragmented populations of greater prairie-chickens. Mol Ecol 12: 3335–3347.
Jordan MA, Snell HL . (2008). Historical fragmentation of islands and genetic drift in populations of Galapagos lava lizards (Microlophus albemarlensis complex). Mol Ecol 17: 1224–1237.
Keller LF, Jeffery KJ, Arcese P, Beaumont MA, Hochachka WM, Smith JN et al. (2001). Immigration and the ephemerality of a natural population bottleneck: evidence from molecular markers. P Roy Soc Lond B Bio 268: 1387–1394.
Knutson MG, Richardson WB, Reineke DM, Gray BR, Parmelee JR, Weick SE . (2004). Agricultural ponds support amphibian populations. Ecol Appl 14: 669–684.
Kramer AT, Ison J, Ashley MV, Howe HF . (2008). The paradox of forest fragmentation genetics. Conserv Biol 22: 878–885.
Kuo CH, Janzen FJ . (2004). Genetic effects of a persistent bottleneck on a natural population of ornate box turtles (Terrapene ornata. Conserv Genet 5: 425–437.
Lande R . (1988). Genetics and demography in biological conservation. Science 241: 1455–1460.
Lande R . (1994). Risk of population extinction from fixation of new deleterious mutations. Evolution 48: 1460–1469.
Li Y . (1993) The General Laws of Species Diversity and Loss for Mammalian and Amphibians on Zhoushan Archipelago and Design Principles for Natural Reserves. Institute of Zoology: Beijing, China.
Lian FQ, Xiao DX . (2001). Studies on the chromosome karyotype of Loropetalum chinense var. rubrum and Loropetalum chinense var. semperrubrum. Acta Agri Univ Jiangxiensis 23: 228–230.
Liu M, Zhang J, Chen Y, Compton SG, Chen XY . (2013). Contrasting genetic responses to population fragmentation in a coevolving fig and fig wasp across a mainland-island archipelago. Mol Ecol 22: 4384–4396.
Luikart G, Allendorf FW, Cornuet JM, Sherwin WB . (1998). Distortion of allele frequency distributions provides a test for recent population bottlenecks. J Hered 89: 238–247.
Luikart G, Cornuet JM . (1998). Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv Biol 12: 228–237.
Luo ZR, Yu MJ, Chen DL, Wu YG, Ding BY . (2012). Spatial associations of tree species in a subtropical evergreen broad-leaved forest. J Plant Ecol 5: 346–355.
Nei M, Maruyama T, Chakraborty R . (1975). The bottleneck effect and genetic variability in populations. Evolution 29: 1–10.
NGDCNH. (2012) The Zhoushan Archipelago. The Navigation Guarantee. The Navigation Guarantee Department (NGD) of the Chinese Navy Headquarter: Tianjin, China.
Petit RJ, El Mousadik A, Pons O . (1998). Identifying populations for conservation on the basis of genetic markers. Conserv Biol 12: 844–855.
Piry S, Luikart G, Cornuet JM . (1999). Bottleneck: a computer program for detecting recent reductions in the effective size using allele frequency data. J Hered 90: 502–503.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Rice WR . (1989). Analyzing tables of statistical tests. Evolution 43: 223–225.
Schaal BA, Leverich WJ . (1996). Molecular variation in isolated plant populations. Plant Spec Biol 11: 33–40.
Spear SF, Peterson CR, Matocq M, Storfer A . (2006). Molecular evidence for historical and recent population size reductions of tiger salamanders (Ambystoma tigrinum in Yellowstone National Park. Conserv Genet 7: 605–611.
Spiegelhalter DJ . (2002). Bayesian measures of model complexity and fit. J Roy Stat Soc B 64: 583–639.
Srikwan S, Woodruff DS . (2000). Genetic erosion in isolated small-mammal populations following rainforest fragmentation. In: Young AG, Clarke GM (eds) Genetics, Demography and Viability of Fragmented Populations. Cambridge University Press: Cambridge. pp 149–172.
Stankowski S, Johnson MS . (2014). Biogeographic discordance of molecular phylogenetic and phenotypic variation in a continental archipelago radiation of land snails. BMC Evol Biol 14: 2.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P . (2004). MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538.
Wang JT, Wang PX . (1980). The relationship between sea level rising and climate change in East China. Chinese J Geographical Sci 35: 299–313. [in Chinese with English abstract].
Wang S, Zhu W, Gao X, Li X, Yan S, Liu X et al. (2014). Population size and time since island isolation determine genetic diversity loss in insular frog populations. Mol Ecol 23: 637–648.
Wang Y, Zhang J, Feelfy KJ, Jiang P, Ding P . (2009). Life-history traits associated with fragmentation vulnerability of lizards in the Thousand-Island Lake, China. Anim Conser 12: 329–337.
Weir BS, Cockerham CC . (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
White TA, Searle JB . (2007). Genetic diversity and population size: island populations of the common shrew, Sorex araneus. Mol Ecol 16: 2005–2016.
Whittaker RJ, Fernández-Palacios JM . (2007) Island Biogeography: Ecology, Evolution, and Conservation. Oxford University Press: Oxford, UK.
Wilson GA, Rannala B . (2003). Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163: 1177–1191.
Wright S . (1978) Evolution and the Genetics of Populations: Variability Within and Among Natural Populations. University of Chicago Press: Chicago, USA.
Young A, Boyle T, Brown A . (1996). The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11: 413–418.
Yuan N, Comes HP, Mao YR, Qi XS, Qiu YX . (2012). Genetic effects of recent habitat fragmentation in the Thousand-Island Lake region of southeast China on the distylous herb Hedyotis chrysotricha (Rubiaceae). Am J Bot 99: 1715–1725.
Zhang X, Pan D, Chen J, Chen P, Jia Y . (2013). Evaluation of land ecological environment in Zhoushan Islands by remotely sensed impervious surfaces. In: Neale CMU, Maltese A (eds) Remote Sensing for Agriculture, Ecosystems, and Hydrology XV. International Society for Optics and Photonics: Dresden, Germany, pp 88871F.
Zhang ZY, Chang HT, Endress PK . (2003). Hamamelidaceae. In: Wu ZY, Raven PH (eds) Flora of China vol. 9, Science Press and Missouri Botanical Garden Press: Beijing, China and St Louis, MO, USA. pp 18–42.
Zhou JL . (1987) The Comprehensive Agriculture Programme of Zhoushan City. Zhejiang Peoples Press: Hangzhou, China.
Acknowledgements
We gratefully acknowledge helpful comments made by two anonymous reviewers and the Subject Editor on a previous version of this article. This research was supported by the National Science Foundation of China (nos. 31170200 and 31370241), the Zhejiang Provincial Funds for Distinguished Young Scientists (no. LR02001) and the Fundamental Research Funds for the Central Universities (no. 2011QNA6013).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

Cite this article
Yuan, N., Comes, H., Cao, Y. et al. A comparative study on genetic effects of artificial and natural habitat fragmentation on Loropetalum chinense (Hamamelidaceae) in Southeast China. Heredity 114, 544–551 (2015). https://doi.org/10.1038/hdy.2014.114
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2014.114
This article is cited by
-
Scale-dependent effects of habitat fragmentation on the genetic diversity of Actinidia chinensis populations in China
Horticulture Research (2020)
-
Microsatellite analysis reveals the resilience of genetic diversity within extant populations of three Akebia species to chronic forest fragmentation in China
Plant Ecology (2019)
-
Habitat fragmentation and biodiversity conservation: key findings and future challenges
Landscape Ecology (2016)
