
Abstract
The Réunion grey white-eye, Zosterops borbonicus, a passerine bird endemic to Réunion Island in the Mascarene archipelago, represents an extreme case of microgeographical plumage colour variation in birds, with four distinct colour forms occupying different parts of this small island (2512 km2). To understand whether such population differentiation may reflect low levels of dispersal and gene flow at a very small spatial scale, we examined population structure and gene flow by analysing variation at 11 microsatellite loci among four geographically close localities (<26 km apart) sampled within the distribution range of one of the colour forms, the brown-headed brown form. Our results revealed levels of genetic differentiation that are exceptionally high for birds at such a small spatial scale. This strong population structure appears to reflect low levels of historical and contemporary gene flow among populations, unless very close geographically (<10 km). Thus, we suggest that the Réunion grey white-eye shows an extremely reduced propensity to disperse, which is likely to be related to behavioural processes.
Similar content being viewed by others

Introduction
The spatial scale of population differentiation varies widely among organisms, mostly as a result of lineage-specific variation in the potential for dispersal and gene flow (Slatkin, 1987). Thus, organisms with shorter dispersal distances are able to differentiate at smaller spatial scales than those with longer dispersal distances, leading to increased opportunities for allopatric or parapatric divergence within a given area (Kisel and Barraclough, 2010). The exact impact of gene flow on population divergence depends upon several factors, including the spatial context of selection and also the balance between the strength of divergent selection pressures and the between-population migration rates (Endler, 1973; Lenormand, 2002). However, reductions in gene flow between adjacent populations that are sufficient to enable divergence seem more likely at larger than at smaller spatial scales for organisms with a given dispersal ability.
In relatively mobile organisms like birds, the strength of gene flow is thought to retard or prevent differentiation between neighbouring populations to such an extent that geographic barriers to dispersal and long isolation times are often considered necessary for genetic differentiation to take place (Mayr and Diamond, 2001; Price, 2010). Although many empirical studies provide support to this idea, with low or non-significant differentiation found even in bird species distributed over a broad geographic scale (Kekkonen et al., 2011; Procházka et al., 2011, for some recent examples), there are also a few striking cases of phenotypic and genetic differentiation among passerine and some other bird populations at relatively small spatial scales (for example, De Léon et al., 2010; Milá et al., 2010).
While it is tempting to invoke a role for strong divergent selection in the face of gene flow to explain such cases of population divergence at small spatial scales, it is often difficult to rule out the possibility that dispersal is reduced or absent without gene flow data because the ability to disperse at given distances does not easily predict the efficiency of dispersal movements, that is, the realized gene flow (Slatkin, 1987; Mallet, 2001), even in birds. For instance, the Seychelles Warbler (Acrocephalus sechellensis) shows locomotory structures similar to those found in closely related species that can sustain flight over long distances, but does not manage to disperse successfully to islands with suitable habitats just outside its distribution range (Komdeur et al., 2004). Thus, to investigate the causes of population divergence at a small spatial scale, it is important to be able to tease apart the effects of gene flow from those due to selection and drift at the relevant spatial scale.
One approach that could be potentially useful relies upon comparing populations of a species in which phenotypic and genetic differentiation occur at a small spatial scale relative to dispersal ability over a range of geographical distances, while minimising the strength of divergent selection pressures by sampling populations experiencing a similar and continuously distributed environment. This should enable, in principle, estimations of dispersal and gene flow independently of the effects of geographic barriers and ecological differences on the patterns of genetic structure.
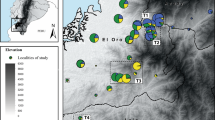
In this study, we use this approach in combination with genetic indirect approaches to investigate the patterns of population genetic structure at a small spatial scale in the Réunion grey white-eye (Zosterops borbonicus). This species complex, endemic to the small island of Réunion (2512 km2), represents an extreme case of microgeographical variation in birds, with parapatrically distributed plumage colour forms restricted to different parts of the island (Gill, 1973; Figure 1). A previous study has shown substantial genetic differentiation among localities distributed across the island, including pairs of localities sampled within the range of the different forms (overall FST analogue for dominant AFLP markers: ΦPT=0.148) (Milá et al., 2010). However, little is known about the evolutionary mechanisms underlying phenotypic and genetic divergence, and no direct or indirect measures of dispersal movements are available for this species. Here we aim to test if restriction of gene flow could have played a role in generating the patterns of genetic differentiation, which have been observed at a very small spatial scale in the Réunion grey white-eye. In order to control for the effects of geographic barriers and ecological differences on population differentiation, we obtained estimates of gene flow from measures of genetic differentiation among localities sampled within the distribution range of one of the colour form, the brown-headed brown form (see description in the studies by Gill (1973) and Milá et al. (2010)), which is entirely restricted to the lower western slopes of Réunion.

Map of Réunion, with sample localities (black dots) and the geographical distribution of the four Réunion grey white-eye plumage colour forms. The geographical coordinates of sample localities are given in Supplementary Table S5. A full color version of this figure is available at the Heredity journal online.
Materials and Methods
Bird samples
We sampled a total of 67 individuals at four sites (Figure 1), where the brown-headed brown form of the Réunion grey white-eye is abundant and broadly distributed across the entire area from low to intermediate elevations. All four sites were located in the central part of the form’s distribution range in a habitat type classified as semi-dry sclerophyllous forest (Thébaud et al., 2009). They were fairly close to one another (mean=16.3 km, ranging from 8.8 to 25.2 km), with no obvious physical barriers to gene flow between them. Field procedures and authorizations have been described elsewhere (Milá et al., 2010).
Molecular procedures
We extracted genomic DNA from blood samples using the DNeasy Blood & Tissue Kit (Qiagen, Venlo, Netherlands). All 67 individuals were genotyped at 12 polymorphic microsatellite loci previously isolated in the study species (Bertrand et al., 2012). PCR amplifications were performed in three 10-μl multiplexes (see Supplementary Table S1), each containing ∼5–30 ng of DNA, 0.2 mM dNTPs, 0.5 μM of each primer and 0.25 U Taq polymerase in 1 × manufacturer’s buffer (2 mM MgCl2). PCR thermal profiles were as follows: initial denaturation at 94 °C for 3 min, followed by 35 cycles at 94 °C for 30 s, locus-specific annealing temperature (see Supplementary Appendix 1) for 30 s, 72 °C for 30 s and a final elongation step at 72 °C for 10 min. Fluorescently labelled PCR products were mixed with formamide. Fragment analysis was carried out on an ABI PRISM 3730 DNA analyser (Applied Biosystems, Foster City, CA, USA) with GeneScan-500(LIZ) size standard. Genotyping profiles were scored using GENEMAPPER v.4.0 software (Applied Biosystems).
Basic genetic analyses and within-population diversity
The presence of null alleles was tested with MICRO-CHECKER v.2.2.3 (van Oosterhout et al., 2004) by running 10 000 Monte Carlo simulations and calculating 95% confidence intervals. The probability of null alleles was negligible for all loci except one (Z16), so we excluded this locus from all analyses. We used GENEPOP v4.0 (Rousset, 2008) to test whether each locus significantly deviated from Hardy–Weinberg equilibrium or showed linkage disequilibrium. Genetic diversity was characterised by calculating the mean number of alleles per locus (A), expected and observed heterozygosity (HE and HO) and FIS values in GENODIVE v2.0 (Meirmans and Van Tienderen, 2004). The allelic richness (AR) corrected for sample size was estimated in FSTAT v2.9.3.2 (Goudet, 2001). To account for potential bias due to family sampling, we examined each population sample for the presence of full-sibs with the COLONY v2.0.4.4 programme (Jones and Wang, 2010). The procedure consisted in four ‘long runs’ with mating system models allowing for polygamy (for both males and females) and inbreeding. We made no further assumption about sibship prior.
Testing for departures from mutation/drift and migration drift equilibriums
Departures from mutation/drift and migration/drift equilibriums might indicate the action of a particular phenomenon (for example, population size variations or restriction in gene flow) so we used two different methods to investigate the likelihood of such departures in our data set. In order to evaluate the possibility for ‘recent’ bottleneck events, departure from mutation/drift equilibrium was tested by comparing levels of observed and expected heterozygosities with the programme BOTTLENECK v1.2.02 (Cornuet and Luikart, 1996; Piry et al., 1999). As the mutation model underlying our microsatellite markers was uncertain, we considered two alternative mutational models: the Stepwise Mutational Model and the Two-phase Model. As recommended by the authors we assumed 95% of single-step mutation for the latter model (Piry et al., 1999). Calculation was run for 10 000 iterations. Wilcoxon tests were then used to estimate whether potential heterozygote excess or deficit were significantly associated with a recent reduction in effective population size. To appreciate the contribution of neutral genetic drift (associated with a low level of migration) in this system, deviation from migration/drift equilibrium was tested by comparing the relative probabilities of a ‘gene flow/drift’ and a ‘drift only’ model with the programme 2MOD (Ciofi et al., 1999). We ran four independent runs setting the MCMC to 1 000 000 iterations (100 000 states burned-in). This method assumes that no allele appeared by mutation since the current population was founded and compares the relative probabilities of the two alternative models.
Population genetic differentiation and gene flow
Data from 11 microsatellite loci were used to estimate levels of genetic differentiation between pairs of populations and among all populations. To obtain indirect estimates of gene flow and compare these estimates with those obtained in other species, we calculated Wright’s fixation indices (FST) using Weir and Cockerham’s estimators θST (1984) as well as a nearly bias-corrected estimator: θRH′ (Raufaste and Bonhomme, 2000), which is particularly suited to weakly differentiated populations. Computations were performed in GENETIX v.4.03 (Belkhir et al., 2004). To enable comparisons with the literature, we also computed GST (Nei, 1987) as well as G″ST (Meirmans and Hedrick, 2011) as implemented in GENODIVE v2.0 (Meirmans and Van Tienderen, 2004). The alternative Dest (Jost, 2008) (also implemented in GENODIVE v2.0) was also computed because its assumptions differ from FST estimators that are under debate in the literature.
We also used the Bayesian multilocus genotyping method implemented in the software BAYESASS 1.3 (Wilson and Rannala, 2003) to detect recent gene flow (over the last several generations) among populations. Three runs of 10 000 000 generations (with a burn-in period of 2 500 000 states) were conducted with all other parameters set to default. This method has been shown to perform well for low migration rate (<33% of migrant individuals per generation) and under moderate genetic differentiation (FST⩾0.05) (Faubet et al., 2007).
Clustering analyses
Bayesian clustering analyses were performed with STRUCTURE 2.3 (Pritchard et al., 2000; Falush et al., 2003) to infer the likelihood of K=1–5 populations. MCMC iterations were set to 500 000 (burn-in period was set to 100 000) with 20 replicates per K. The optimal value of K was evaluated by considering the highest mean likelihood value of K, that is, L(K), as well as the ΔK method (Evanno et al., 2005). The optimal alignment of the 20 replicates was determined with the Greedy algorithm implemented in CLUMPP (Jakobsson and Rosenberg, 2007). All analyses were run using the admixture model (and correlated allele frequencies), which provides us with estimates of admixture proportions for each individual among clusters. We also used the LOCPRIOR model implemented in STRUCTURE 2.3, as it is more efficient at detecting genetic structure at lower levels of divergence than previous STRUCTURE models. This model makes use of information about sampling locations, but it does not tend to detect any sub-structure when none is present and will ignore sample group information when the ancestry of individuals does not correlate with sampling locations (Hubisz et al., 2009).
Results
Within-population genetic diversity and equilibriums
All microsatellite loci were in Hardy–Weinberg equilibrium. No significant linkage disequilibrium was found across all pairs of loci after correcting for multiple comparisons with the sequential Bonferonni procedure (Rice, 1989). All populations presented similar levels of intra-population polymorphism (with A ranging from 6.27–8.09 and AR ranging from 6.09–6.39) (Table 1). Within-population tests of mutation/drift equilibrium provided no evidence of heterozygote excess (P>0.18) for both the Two-Phase model and the Stepwise Mutational model (Table 2). Bayes factors comparing the models of ‘pure drift’ versus ‘gene flow-drift’ models were close to 8, strongly supporting the ‘pure drift’ over the ‘gene flow/drift’ model (Table 3). This result alone suggests that gene flow is significantly restricted between populations.
Among-population genetic differentiation and clustering
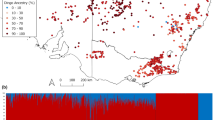
The programme COLONY 2.0.4.4 identified two fullsib pairs in one locality (St–Leu) and a group of four individuals in a second locality (Canot). We ran the analyses twice with either the full data set or with a reduced data set from which one randomly chosen individual from each fullsib pair and three randomly chosen individuals within the group of four were excluded. The results we obtained in the two sets of analyses are qualitatively and quantitatively similar (see Table 4 and Supplementary Table S3 and S4), and so we present only the results using the full data set. Overall estimates of among-population genetic differentiation were low but significant for all indices (θST≈GST=0.03, θRH′=0.22, G″ST=0.14, Dest=0.12, P<0.05) (see Supplementary Table S1). All pairwise comparisons were also significant (Table 4), independent of the geographic distance between sample localities. Although there were differences in the magnitude of genetic differentiation between indices, the global pattern of pairwise differentiation was consistent across indices (Table 4). We also found that the most likely number of genetic clusters is three (K=3, L(K)=−2902.635, ΔK=9.640), with all individuals unambiguously assigned to the different clusters (Figure 2, see also Supplementary Table S2). The first two clusters matched exactly two sample localities, whereas the third one consisted of individuals from the two sample localities in close geographic proximity to one another. Thus, sample localities are broadly differentiated from one another at a very small spatial scale (<100 km2), likely as a result of limited gene exchange among them. However, even at that scale, reductions in gene flow between adjacent sample localities appear more likely at larger than at smaller spatial scales.

Admixture proportions as inferred from genetic clustering. Each bar represents an individual. Each colour reflects the likelihood of belonging to one of the inferred genetic clusters. Dashed lines delimit sample localities.
Contemporary gene flow
Most sample localities have low proportion of migrant (<9%) and high proportions of non-migrant (>88%) individuals per generation, with the exception of the sites situated at close geographic proximity to its nearest neighbour (8.8 km), which seems to have a large expected proportion of migrants (22%). We found no evidence of asymmetry in migration between adjacent localities and among all localities (Table 5). Thus, these results are consistent with the idea that gene flow and dispersal are extremely reduced between adjacent localities, unless very close (<10 km).
Discussion
Levels of population genetic differentiation implies reduced dispersal and gene flow
We found a significant signal of population differentiation across four sample localities situated within the distribution range of the brown-headed brown form of the Réunion grey white-eye. Accordingly, we demonstrated the presence of population structure by differentiating three clusters of individuals within the data set, with the two sample localities situated within 10 km of each other forming one of the clusters. This shows that population structuring in the Réunion grey white-eye can occur at a scale of 10–20 km even in a broadly continuous habitat. The levels of genetic differentiation observed in this study are unexpectedly high, given the spatial scale considered. While comparing the magnitude of genetic differentiation found in different studies is not devoid of problems (for example, Meirmans and Hedrick, 2011; Whitlock, 2011), our estimates (θST≈GST=0.03=, θRH′=0.22, G″ST=0.14, Dest=0.12) are close to average FST values reported in previous studies (0.049 in the study by Barrowclough (1983) and 0.048 in the study by Evans (1987)). It is also striking that these estimates are comparable to those found at the scale of much larger regions in other passerines such as the House Sparrow (Passer domesticus) (FST=0.004 across Finland; Kekkonen et al., 2011) or the Eurasian Reed Warblers (Acrocephalus scirpaceus) (FST=0.013, G′ST=0.078, Dest=0.063 across Europe; Procházka et al., 2011). Thus, we suggest that the levels of genetic differentiation found in our study are not just unexpected, they are exceptionally high for birds at such a small spatial scale.
Both significant levels of genetic differentiation and population structure are therefore consistent with extremely reduced gene flow between populations. It is noteworthy that our comparison of ‘pure drift’ and the ‘gene flow/drift’ models also supports the idea that drift is prevalent relative to migration in explaining differentiation. Our analysis of contemporary gene flow also suggests that current levels of gene flow among populations are very low, in agreement with our suggestion that historical gene flow must have been extremely reduced to explain present-day patterns of genetic differentiation.
Genetic differentiation with no geographic and ecological transitions
We compared sample localities that are very close to one another, occupy a similar habitat type and are not separated by any obvious physical or ecological discontinuities. Thus, our results likely reflect reduced dispersal and gene flow at a very small spatial scale, independently of the effects of divergent selection pressures and geographic barriers to gene flow.
There are several explanations for such an unusual pattern of genetic differentiation at a small scale. First, we assumed that the populations were experiencing similar environments, using habitat type as a proxy. Environmental factors, including for example, altitude, temperature or rainfall, often vary within a habitat type, especially on islands with a very rugged topography (Whittaker and Fernandez-Palacios, 2007). In addition, biotic factors such as parasites and pathogens may also vary between localities within a same habitat type. Populations could then diverge in response to heterogeneous natural selection, and this may keep gene flow at low levels if immigrants have reduced fitness relative to residents. This was found in wild populations of great tits (Parus major) separated by distances of <3 km (Garant et al., 2005; Shapiro et al., 2006; Björklund et al., 2010), and also in song sparrows (Melospiza melodia) with five subspecies coexisting in a restricted area made of various microhabitats (Chan and Arcese, 2003). However, the latter results were not associated with significant neutral genetic differentiation, which suggests that genetic drift was not strong enough to generate neutral genetic differentiation (but also refer to the studies by Senar et al. (2006), Lee et al. (2010) and Rutz et al. (2012)). There is no evidence from previous studies that populations belonging to one particular colour form of the Réunion grey white-eye and being as geographically close as those used in this study show any sign of niche differentiation (Gill, 1971, 1973), suggesting that this explanation is unlikely.
Subtle, undetected geographic barriers could also account for reduced gene flow between populations. However, this seems also unlikely as effective geographic barriers for the Réunion grey white-eye are conspicuous physiographic features such as major river beds and extensive lava flows (Gill, 1973; Milá et al., 2010), none of which occur in our study area. A more plausible explanation is that the Réunion grey white-eye, like some other island birds including several species of white-eyes, could show a reduced propensity to disperse, perhaps as a result of selection against long-distance dispersal (Komdeur et al., 2004; Moyle et al., 2009). As the species is clearly capable of sustained flight over hundreds of metres of land, it may express the phenomenon of ‘behavioural flightlessness’, that is, a behavioural reluctance to move away from its source locality (Diamond, 1981). Very high recapture rates on long-term study sites could be consistent with this idea (Milá and Thébaud, unpublished data).
The extent to which social behaviour could also influence the spatial genetic structure of populations is largely unknown (Painter et al., 2000), but substantial levels of genetic differentiation was found at relatively small spatial scales in lekking (Höglund and Shorey, 2003; Bouzat and Johnson, 2004) or cooperative breeding bird species (Painter et al., 2000; Double et al., 2005; Temple et al., 2006; Woxvold et al., 2006). Strong social behaviours such as allopreening, huddling and cooperative breeding are common in the Réunion grey white-eye, with no apparent territorial behaviour throughout the breeding season (Gill, 1971; Gill, 1973), and may further contribute to reducing gene exchange among populations located at very short distances (for example, through social structuring and/or strong philopatry). Clearly, more work needs to be done to understand if such behavioural processes can be associated with reductions in gene flow among populations.
Another possible explanation relies on the idea that vocal microgeographic variation in the form of song ‘dialects’ may easily arise in birds that learn their songs (Catchpole and Slater 2008), such as white-eyes (Baker 2012). This could potentially contribute to reductions in gene flow among geographically close populations. If neighbouring populations differ in their song types, with young males and females preferentially learning local song types and then, later in life, preferring these songs while discriminating against non-local variants, then interpopulation matings could be reduced relative to intrapopulation matings, causing a restriction in gene flow (MacDougall-Shackleton and MacDougall-Shackleton, 2001). This seems especially likely in resident species, but it will ultimately depend upon natal dispersal distances. In the case of the Réunion grey white-eye, whether vocal dialects may contribute to population differentiation has yet to be tested.
We have shown that populations of the Réunion grey white-eye can exhibit spatial genetic structure and differentiation at a very small scale (<100 km2), even in the absence of any obvious geographic barrier and/or change in habitat attributes. This strong population structure appears to reflect low levels of historical and contemporary gene flow among populations, unless very close geographically (<10 km). Thus, the Réunion grey white-eye seems to show an extremely reduced propensity to disperse, which is likely to be related to behavioural processes, because the birds show no sign of wing reduction or of a reduced power of flight. Besides the fact that the pattern seen here reveals levels of genetic differentiation at a small spatial scale, which are exceptionally high for birds that are good flyers, our findings also have implications for how the different colour forms found in the Réunion grey white-eye have likely been shaped by the interplay of natural selection, genetic drift and reduced gene flow.
Data archiving
Data deposited in the Dryad repository: doi:10.5061/dryad.1652m.
References
Baker MC . (2012). Silvereyes (Zosterops lateralis) song differentiation in an island-mainland comparison: analyses of a complex cultural trait. Wilson J Ornithol 124: 454–466.
Barrowclough GF . (1983). Biochemical studies of microevolutionary processes. In: Brush AH, Clark GA Jr (eds) Perspectives in Ornithology. University of Cambridge Press: Cambridge, UK, pp 223–261.
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F . (2004). GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. Laboratoire Génome Populations Interactions CNRS UMR5 117: 5000.
Bertrand JAM, García-Jiménez R, Bourgeois Y, Duval T, Heeb P, Thébaud C et al. (2012). Isolation and characterization of twelve polymorphic microsatellite loci for investigating an extreme case of microgeographical variation in an island bird (Zosterops borbonicus). Conservation Genet Res 4: 323–326.
Björklund M, Ruiz I, Senar JC . (2010). Genetic differentiation in the urban habitat: the great tits (Parus major) of the parks of Barcelona city. Biol J Linn Soc 99: 9–19.
Bouzat J, Johnson K . (2004). Genetic structure among closely spaced leks in a peripheral population of lesser prairie-chicken. Mol Ecol 13: 499–505.
Catchpole CK, Slater PJB . (2008) Bird Songs: Biological Themes and Variations. Cambridge University Press: Cambridge, UK.
Chan Y, Arcese P . (2003). Morphological and microsatellite differentiation in Melospiza melodia (Aves) at a microgeographic scale. J Evol Biol 16: 939–947.
Ciofi C, Beaumont MA, Swingland I R, Bruford MW . (1999). Genetic divergence and units for conservation in the Komodo dragon Varanus komodoensis. Proc R Soc B Biol Sci 266: 2269–2274.
Cornuet JM, Luikart G . (1996). Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144: 2001–2014.
De Léon LF, Bermingham E, Podos J, Hendry AP . (2010). Divergence with gene flow as facilitated by ecological differences: within-island variation in Darwin’s finches. Phil. Trans. R. Soc. B 365: 1041–1052.
Diamond JM . (1981). Flightlessness and fear of flying in island species. Nature 293: 507–508.
Double MC, Peakall R, Beck NR, Cockburn A . (2005). Dispersal, phylopatry, and infidelity: dissecting local genetic structure in superb fairy-wrens (Malurus Cyaneus). Evolution 59: 625–635.
Endler JA . (1973). Gene flow and population differentiation. Science 179: 243–250.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Evans PGH . (1987). Electrophoretic variability of gene products. In: Cooke F, Buckley PA (eds) Avian Genetics. Academic Press: New York, NY, USA. pp 105–162.
Falush D, Stephens M, Pritchard JK . (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164: 1567–1587.
Faubet P, Waples RS, Gaggiotti OE . (2007). Evaluating the performance of a multilocus Bayesian method for the estimation of migration rates. Mol Ecol 16: 1149–1166.
Garant D, Kruuk LEB, Wilkin TA, McCleery RH, Sheldon BC . (2005). Evolution driven by differential dispersal within a wild bird population. Nature 433: 60–65.
Gill FB . (1971). Ecology and evolution of the sympatric Mascarene white-eyes, Zosterops borbonica and Zosterops olivacea. Auk 88: 35–60.
Gill FB . (1973). Intra-island variation in the Mascarene White-eye Zosterops borbonica. Ornithol Monogr 12: 1–66.
Goudet J . (2001). FSTAT, a program to estimate and test gene diversities and fixation indices. Version 2: 3. J Heredity 86: 485–486.
Höglund J, Shorey L . (2003). Local genetic structure in a white-bearded manakin population. Mol Ecol 12: 2457–2463.
Hubisz MJ, Falush D, Stephens M, Pritchard JK . (2009). Inferring weak population structure with the assistance of sample group information. Mol Ecol Res 9: 1322–1332.
Jakobsson M, Rosenberg NA . (2007). CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23: 1801–1806.
Jones OR, Wang J . (2010). COLONY: a program for parentage and sibship inference from multilocus genotypes data. Mol Ecol Res 10: 551–555.
Jost L . (2008). GST and its relatives do not measure differentiation. Mol Ecol 17: 4015–4026.
Kekkonen J, Seppä P, Hanski IK, Jensen H, Väisänen RA, Brommer JE . (2011). Low genetic differentiation in a sedentary bird: house sparrow population genetics in a contiguous landscape. Heredity 106: 183–190.
Kisel Y, Barraclough TG . (2010). Speciation has a spatial scale that depends on level of gene flow. Am Nat 175: 316–334.
Komdeur J, Piersma T, Kraiijeveld K, Kraiijeveld-Smit F, Richardson DS . (2004). Why Seychelles warblers fail to recolonize nearby islands: unwilling or unable to fly there? Ibis 146: 298–302.
Lee JW, Simeoni M, Burke T, Hatchwell BJ . (2010). The consequences of winter flock demography for genetic structure and inbreeding risk in vinous-throated parrotbills, Paradoxornis webbianus. Heredity 104: 472–481.
Lenormand T . (2002). Gene flow and the limit to natural selection. Trends Ecol Evol 17: 183–189.
MacDougall-Shackleton EA, MacDougall-Shackleton SA . (2001). Cultural and genetic evolution in mountain White-crowned Sparrows: song dialects are associated with population structure. Evolution 55: 2568–2575.
Mallet J . (2001). Gene flow. In: Woiwod IP, Reynolds DR, Thomas CD (eds) Insect Movement: Mechanisms and Consequences. CAB International. pp 337–360.
Mayr E, Diamond JM . (2001) The Birds of Northern Melanesia: Speciation, Ecology, and Biogeography. Oxford University Press: Oxford, UK.
Meirmans PG, Hedrick PW . (2011). Assessing population structure: FST and related measures. Mol Ecol Res 11: 5–18.
Meirmans PG, Van Tienderen PH . (2004). Genotype and Genodive: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4: 792–794.
Milá B, Warren BH, Heeb P, Thébaud C . (2010). The geographic scale of diversification on islands: genetic and morphological divergence at a very small spatial scale in the Mascarene grey white-eye (Aves: Zosterops borbonicus). BMC Evol Biol 10: 158.
Moyle RG, Filardi CE, Smith CE, Diamond J . (2009). Explosive Pleistocene diversification and hemispheric expansion of a ‘great speciator’. Proc Natl Acad Sci USA 106: 1863–1868.
Nei M . (1987) Molecular Evolutionary Genetics, MacIntyre RJ (eds) Columbia University Press: New York, NY, USA.
Painter J, Crozier R, Poiani A, Robertson R, Clarke M . (2000). Complex social organization reflects genetic structure and relatedness in the cooperatively breeding bell miner, Manorina melanophrys. Mol Ecol 9: 1339–1347.
Piry S, Luikart G, Cornuet JM . (1999). BOTTLENECK: A computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90: 502–503.
Price TD . (2010) Speciation in Birds. Roberts and Company Publishers: Greenwood Village, CO, UK.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Procházka P, Stokke BG, Jensen H, Fainová D, Bellinvia E, Fossøy F et al. (2011). Low genetic differentiation among reed warbler Acrocephalus scirpaceus populations across Europe. J Avian Biol 42: 103–113.
Raufaste N, Bonhomme F . (2000). Properties of bias and variance of two multiallelic estimators of FST . Theor Popul Biol 57: 285–296.
Rice WR . (1989). Analyzing tables of statistical tests. Evolution 43: 223–225.
Rousset F . (2008). genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Res 8: 103–106.
Rutz C, Ryder TB, Fleischer RC . (2012). Restricted gene flow and fine-scale population structuring in tool using New Caledonian crows. Naturwissenschaften 99: 313–320.
Senar JC, Borras A, Cabrera J, Cabrera T, Björklund M . (2006). Local differentiation in the presence of gene flow in the citril finch Serinus citrinella. Biol Lett 2: 85–87.
Shapiro BJ, Garant D, Wilkin TA, Sheldon BC . (2006). An experimental test of the causes of small-scale phenotypic differentiation in a population of great tits. J Evol Biol 19: 176–183.
Slatkin M . (1987). Gene flow and the geographic structure of natural populations. Science 236: 787–792.
Thébaud C, Strasberg D, Warren BH, Cheke A . (2009). Mascarene islands, biology. In: Gillespie RG, Clague DA (eds) Encyclopedia of Islands. University of California Press: Berkeley, CA, USA, pp 612–619.
Temple HJ, Hoffman JI, Amos W . (2006). Dispersal, philopatry and intergroup relatedness: fine-scale genetic structure in the white-breasted thrasher, Ramphocinclus brachyurus. Mol Ecol 15: 3449–3458.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P . (2004). Micro-Checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538.
Weir B, Cockerham CC . (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Whitlock MC . (2011). G’ST and D do not replace FST . Mol Ecol 20: 1083–1091.
Wilson GA, Rannala B . (2003). Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163: 1177–1191.
Whittaker RJ, Fernandez-Palacios JM . (2007) Island biogeography: ecology, evolution and conservation. Oxford University Press: Oxford, UK.
Woxvold IA, Adcock GJ, Mulder RA . (2006). Fine-scale genetic structure and dispersal in cooperatively breeding apostlebirds. Mol Ecol 15: 3139–3146.
Acknowledgements
We thank the editor and two anonymous referees for their insightful and constructive comments that improved an earlier version of this manuscript. Fieldwork was facilitated by the outstanding efforts of Guillaume Gélinaud, Dominique Strasberg, Juli Broggi, Ben Warren, Magali Thierry, René-Claude Billot, Jean-Michel Probst, Isabelle Henry and Vincent Leconte. We gratefully thank the Réunion National Park for permission to conduct fieldwork. Marc Salamolard and Benoît Lequette provided valuable help with fieldwork and logistics. JB, YB, BD and JC were supported by MESR (Ministère de l’Enseignement Supérieur et de la Recherche) PhD scholarships. The research was supported by Agence Française pour le Développement grants to CT, the Fondation pour la Recherche sur la Biodiversité (FRB) through its Centre for Synthesis and Analysis of Biodiversity (CESAB), the ‘Laboratoire d’Excellence’ TULIP (ANR-10-LABX-41) and the SYNTHESYS Project (http://www.synthesys.info/), which was funded by the European Community Research Infrastructure Action under the FP7 ‘Capacities’ Programme at the Museo Nacional de Ciencias Naturales (CSIC) of Madrid, Spain.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Bertrand, J., Bourgeois, Y., Delahaie, B. et al. Extremely reduced dispersal and gene flow in an island bird. Heredity 112, 190–196 (2014). https://doi.org/10.1038/hdy.2013.91
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2013.91
Keywords
This article is cited by
-
Habitat fragmentation differentially shapes neutral and immune gene variation in a tropical bird species
Heredity (2021)
-
Adult survival selection in relation to multilocus heterozygosity and body size in a tropical bird species, the Zenaida dove, Zenaida aurita
Oecologia (2016)
-
Survival and dispersal of the Cyprus wheatear Oenanthe cypriaca, an endemic migrant
Journal of Ornithology (2016)
-
Contrasted patterns of genetic differentiation across eight bird species in the Lesser Antilles
Genetica (2016)
-
The evolution of contact calls in isolated and overlapping populations of two white-eye congeners in East Africa (Aves, Zosterops)
BMC Evolutionary Biology (2014)
