
Abstract
The Bedouin Israeli population is highly inbred and structured with a very high prevalence of recessive diseases. Many studies in the past two decades focused on linkage analysis in large, multiple consanguineous pedigrees of this population. The advent of high-throughput technologies motivated researchers to search for rare variants shared between smaller pedigrees, integrating data from clinically similar yet seemingly non-related sporadic cases. However, such analyses are challenging because, without pedigree data, there is no prior knowledge regarding possible relatedness between the sporadic cases. Here, we describe models and techniques for the study of relationships between pedigrees and use them for the inference of tribal co-ancestry, delineating the complex social interactions between different tribes in the Negev Bedouins of southern Israel. Through our analysis, we differentiate between tribes that share many yet small genomic segments because of co-ancestry versus tribes that share larger segments because of recent admixture. The emergent pattern is well correlated with the prevalence of rare mutations in the different tribes. Tribes that do not intermarry, mostly because of social restrictions, hold private mutations, whereas tribes that do intermarry demonstrate a genetic flow of mutations between them. Thus, social structure within an inbred community can be delineated through genomic data, with implications to genetic counseling and genetic mapping.
Similar content being viewed by others

Introduction
The presence of a structure within a population is one of the main challenges in the design of population genetic studies (Price et al., 2010; Astle and Balding, 2009; Yu et al., 2005; Sun et al., 2002; Mathieson and McVean, 2012). Whereas efficient methods have been developed to detect variations between large, divergent populations, the fine structure patterns within populations are often too subtle to be captured by current approaches for stratification (Mathieson and McVean, 2012). One of the main approaches for studying fine patterns within populations is by inferring relatedness (Purcell et al., 2007; Gusev et al., 2009; Bercovici et al., 2010; Browning, 2008). A group of individuals that is more related to each other than to other members in the population may potentially hold common risk alleles for both common and rare traits (Mathieson and McVean, 2012; Peltonen et al., 2000). Such constellations could be because of spatial constraints, religious and other social polarization, or traditions linked to common historical events. Examples for such patterns could be found with the Amish (Strauss and Puffenberger, 2009) the French Canadians, and the Bedouin populations (Laberge et al., 2005; Mohammad et al., 2009; Teebi, 1997).
In the current study, we set out to study the relatedness between the different tribes of the Bedouin population in the Negev region of southern Israel. There are two major motivations for this study. The first is to establish a methodology for delineating tribal relatedness and admixture patterns, so that risk factors for rare genetic diseases could be assessed more accurately during genetic counseling and carrier testing (that is, related tribes may hold common risk alleles). Another motivation is to utilize the inferred tribal relatedness for genetic mapping in sporadic cases collected from different tribes. In order to develop a methodology for mapping tribal relatedness, it is instructive to learn about the traditional structure of Bedouin tribes. The Bedouins are nomadic people present throughout the Middle East, most of whom migrated from the Arabian Peninsula (Mohammad et al., 2009; Hunter-Zinck et al., 2010; Sheffield et al., 1998; Marx, 1967; Cavalli-Sforza et al., 1994). The Bedouin social structure is organized in a very hierarchical manner: several basic units of nuclear families are integrated into a tribe (an extended family of relatives), and the federation of several tribes forms a clan (Marx, 1977; Chatty, 2006). Consanguineous marriages within the tribe unit are common. Marriages between the different tribes in a clan are less frequent. Marriages between clans are rare and in many cases forbidden (Mohammad et al., 2009; Marx, 1977). Tribes and clans may vary substantially in size. According to our records for the Negev Bedouins, tribes could be as small as 100 individuals or as large as 10 000 individuals. Furthermore, this population is dynamic in the sense that tribes may converge or diverge as a result of climate or social and political constraints (Al-Aref, 1934). In particular we note that, although surnames generally indicate tribal origin, they are not always accurate indicators for ancestry. Tribes may adapt new surnames without having any blood ties to the hosting tribe or clan.
The Bedouin population of the Negev is a small founder population of ∼200 000 individuals, which, according to our records of surnames, is distributed in well over a hundred tribal units. Historical records indicate that the ancestors of most of the Negev Bedouins migrated from the Arabian Peninsula to southern Israel and Sinai probably before the spread of the Islam around 700 CE (Marx, 1967; Al-Aref, 1934; Abu-Rabia, 1994; Bailey, 1985). These ancient Bedouins formed several clans and established clan territories in the Negev and Sinai deserts. The region has also known a constant flow of small sporadic tribes that migrated to the Negev from Egypt, Jordan and Gaza. These sporadic tribes were allowed to stay in the region by forming alliances with the big clans. Although the sporadic tribes were usually kept as separate entities, not allowed to marry into the hosting clans, there were no restrictions regarding intermarriages between the different sporadic tribes. Indeed, in our field studies we found that small tribes that do not possess kinship to the large clans allow intertribal marriages. A possible explanation to this social structure is the historical use of kinship lineages as a mean to control territory, which is associated with the clan identity (Marx, 1977).
Considering the complex tribal structure just described, it is challenging to interpret kinship in this population. Indeed, genetic mapping in the past two decades revealed rare mutations that were common across tribes, the presence of which may suggest common founder ancestry, or possibly reflect recent admixture between the tribes because of intermarriages. Uncovering those different scenarios through genomic data is one of the main challenges in this study.
Relatedness is often studied by inferring segment-shared Identical By Descent (IBD) from common ancestors (Sun et al., 2002; Purcell et al., 2007; Weir et al., 2006; Browning 2008). Close relatives, separated by just a few generations, are expected to share a small number of very long segments inherited from their most recent common ancestor. Distant relatives, separated by many generations, may share many short segments from several distant ancestors. Thus, the distribution of shared segments between individuals holds information on the unique lineages to common ancestors and thus provides valuable input for the inference of population structure (Huff et al., 2011; Lawson et al., 2012). In many studies, however, IBD inference is related to a single, most recent common ancestor. This is because exact lineages, especially those of the distant past, are difficult to infer, often requiring specific genetic and demographic models and large reference population panels (Gasbarra et al., 2007; Pool et al., 2010; Novembre and Ramachandran, 2011). In isolated populations and in populations with a high degree of consanguinity, the problem is even more challenging because the background level of IBD sharing between unrelated individuals is inflated, not allowing efficient discrimination between recent ancestry and background sharing (Huff et al., 2011).
Our approach to the analysis of fine structure within the Bedouin population is based on a methodology recently suggested by Lawson et al. (2012). In this approach, a similarity matrix is first constructed between all pairs of individuals and then a clustering procedure rearranges the order of the pairs to reveal various patterns and kin groups in the data. Here, we suggest several modifications to their method that better fit the study of structured isolated populations with elevated sharing of recent ancestry. We tackle the ancestry-inference problem with a modification to the existing model for IBD inference implemented in the popular PLINK tool (Purcell et al., 2007). We introduce an algorithm for recent ancestry inference that calculates effective kinship coefficients for each pair in the data. The effective kinship is simply the sum of all detected segments shared by two individuals, without differentiating between different ancestors. We also suggest a metric, the residual kinship, for the detection of co-ancestry, given that recent ancestry is present. The motivation behind the residual ancestry is to differentiate between recent admixture of subpopulations (tribes in our case) and shared co-ancestry that is more ancient. Our algorithm calculates a Maximum Likelihood Estimator for the effective kinship coefficients with no prior genealogical knowledge. We then reorganize the kinship matrix using SPIN, an algorithm designed to reorder distance matrices (Tsafrir et al., 2005). The sorted kinship matrix is then analyzed for fine structure between the different tribes. Applying this methodology to samples from the Negev Bedouin population, we were able to reconstruct the large clans and uncover patterns of tribal interactions that are consistent with the known oral history. We also find a correlation between the patterns of tribal relatedness and rare variants known to be common in related tribes. We discuss the implications of our findings for genetic counseling and IBD mapping in the Bedouin community with specific examples.
Materials and methods
The Human Diversity Genome Project (HDGP) data consist of 45 individuals (kinship coefficient <0.15 as calculated with the Method of Moments kinship in PLINK (Purcell et al., 2007)) of the Negev Bedouin community included in the CEPH Human Genome Diversity Panel. Individuals were previously genotyped with the Illumina HumanHap550 SNP (single-nucleotide length polymorphism) platform at 513 008 biallelic autosomal genetic markers as previously reported by Jakobsson et al. (2008). We selected 96 123 SNPs for the analysis, with minor allele frequency >5% and a median distance of 24 kbp between adjacent markers. The LAB data set consists of 30 individuals from 16 different tribes as indicated in Supplementary Table S1. Genotyping, following informed consent and approval by the Soroka Medical Center IRB, was performed using Affymetrix GeneChip Human Mapping 250K Nsp Arrays, according to the manufacturer’s guidelines. A total of 83 809 SNPs were selected for this analysis, with minor allele frequency >5% and a median distance of 22 kbp between adjacent markers. The LAB data include a number of known relatives to assess the within-pedigree relatedness. A subset of 18 seemingly unrelated individuals was constructed for between-pedigree analyses (Method of Moments kinship coefficient <0.15).
Throughout this paper, we use the term kinship to designate the effective kinship between each pair. The effective kinship is defined here to be the sum of all shared segments inferred to be IBD by a model that assumes an open large population. It coincides with the usual definition of kinship coefficients when a pair of individuals is related by a single lineage to recent ancestors. Thus, in our methodology, a pair of individuals is related by an effective ancestor who is the sum of all recent ancestors linking the pair. This approach is obviously an approximation; however, it substantially simplifies the inference of kinships and seems to satisfy the task of clan identification. The main motivation for using this approximation here is that, within clans, the sharing of recent ancestry between individuals is expected to be much higher than between clans. It is thus expected that by establishing the proportion of sharing, one can differentiate clan members from sporadic sharing in the population. The choice of the Log scale for the dissimilarity scores deserves some explanation as well. In open populations, the dissimilarity metric defined here is related to the number of generations separating the pair (Kirkpatrick et al., 2011). Whereas the interpretation of this metric in our case is more complicated, we chose to retain it mainly because of the observation that the distribution of this metric is approximately normal within and between clans (see Supplementary Figure S1).
Shared regions and kinship coefficients for each pair were inferred using the IBD analysis (Lander and Green, 1987). IBD inference was performed with a modified version of the PLINK (Purcell et al., 2007) tool, which we designate as PLINKem. The modified version involves two algorithmic generalizations to the original pairwise IBD inference method. The first is the incorporation of an error model as previously described by Markus et al. (2011) and implemented in PLINKe. For the Affymetrix arrays (LAB) we used the IBD model that incorporates the confidence scores of the genotyping procedure to account for the different error rates in the data (Markus et al., 2011). For the Illumina arrays (HDGP), we assumed a constant error rate of 1%. The second generalization is more subtle and involves an iterative procedure that aims to find the most likely effective kinship coefficients connecting each pair of individuals. The next paragraphs describe this algorithm in detail.
Pairwise IBD Inference in PLINK is implemented by estimating the conditional probability of IBD states on the marker data. Following the definitions from the PLINK paper, the set of measured markers denoted by M, and let z=0, 1 or 2 be the IBD states of sharing 0, 1 or 2 alleles between the pair, respectively. Let Zi be the IBD state at a particular position i in the genome. The conditional probability of IBD state on marker data is expressed by the Bayes theorem as:

To evaluate this expression, the authors of PLINK defined an HMM with emission and transition matrices in Tables 2 and 3 of their manuscript, respectively (Purcell et al., 2007). In particular, the definition of the transition matrix involves an estimate of the number of meioses between the pair of individuals. This parameter is estimated from the global IBD probabilities for sharing z=0, 1 or 2 alleles anywhere in the genome as indicated in the PLINK manuscript in the Appendix. These global IBD probabilities are estimated from the data using a method of moments, which is essentially an average of the single-locus IBD probabilities. This solution is somewhat artificial in the sense that it is not part of the model given by equation (1).
The method we propose is to estimate the global IBD probabilities from the model itself using maximum likelihood. Denoted by K=(k0,k1,k2), the set of three kinship coefficients such that kz=P(Z=z) is the global probability of sharing z allele IBD between the pair. Let N be the number of chromosomes and Mn the marker data on chromosome n. We can write the likelihood for the kinship coefficients given the genome-wide marker data as follows,

In this expression, PK(Mn) is the probability of the data on chromosome n, given that the kinship coefficients (K) are known. Once the kinship coefficients are given, the calculation of this quantity is similar to that described in the PLINK paper. The goal is then to find the kinship coefficients that maximize this likelihood. This task is difficult, as the state space is made up of all possible combinations of IBD states and thus is exponential in the number of loci. Thus, using the Maximization Expectation (EM) method is not feasible. Instead, we proposed a modified EM that uses the IBD inference of successive iterations for the update of the kinship parameter. The algorithm is described in Appendix A. One important note on this algorithm should be made. At stage three, we can apply filters on the counting of the segments that were inferred to be shared by the pair. For example, when inferring recent ancestry it is common to count segments >3–5 cM, (Huff et al., 2011; Atzmon et al., 2010) which enables control over Linkage Disequilibrium (LD) patterns (Purcell et al., 2007). It also allows us to create different kinship matrices at different ancestry depths as described in Supplementary Appendix B.
In the tests we performed, our algorithm converged after approximately three iterations on average. We compared our method with several other approaches on real data from our laboratory and on simulated data generated with SimPed (Leal et al., 2005) (see Supplementary Appendix A and Supplementary Tables S2 and S3). The other approaches include exact inference using MERLIN, (Abecasis et al., 2001) approximated inference using PREST (Sun et al., 2002) and PLINK Method of Moments (MoM) kinship estimation (genome option). The method of moments used in PLINK assume that all markers are independent, thus trying to estimate the kinship coefficients from each marker separately. This is performed by establishing the probability of observing the shared alleles, given the possible IBD states between two individuals. Averaging over all markers and using the Bayes theorem give a global probability estimate for sharing allele IBD. The method in PREST is more sophisticated. While still assuming the markers to be unlinked, it tries to maximize the likelihood of the kinship coefficients given the data. The difference between PREST and our model is that, in PREST, the likelihood function is based on a single-locus model, whereas our model uses a variation on the multilocus method of Lander and Green (1987). As suggested by the analysis on real data sets (Supplementary Appendix A and Supplementary Figure S3), PLINKem is substantially more accurate than both PREST and MoM estimations. It should be noted that multipoint methods are substantially slower than single-point methods and therefore are restricted to small data sets (typically N<100).
After the establishment of the various kinship matrices, we explored the relationship between individuals by permuting the order of the individuals in the matrices using SPIN (Sorting Points Into Neighborhoods) (Tsafrir et al., 2005). From the inferred kinship coefficients, the average kinship sharing was calculated as  for all pairs. We defined the dissimilarity matrix to be
for all pairs. We defined the dissimilarity matrix to be  and used it as input for the SPIN algorithm. Elements of ρ having a zero value were assigned the smallest non-zero value in the matrix. We permuted Dcombined using the Neighborhood sort, which finds a linear ordering on the individuals such that pairs closer to each other in the permuted matrix also have small dissimilarity values (Tsafrir et al., 2005). We used the ordering from Dcombined to present the other two matrices, Drecent and Dresidual, as well.
and used it as input for the SPIN algorithm. Elements of ρ having a zero value were assigned the smallest non-zero value in the matrix. We permuted Dcombined using the Neighborhood sort, which finds a linear ordering on the individuals such that pairs closer to each other in the permuted matrix also have small dissimilarity values (Tsafrir et al., 2005). We used the ordering from Dcombined to present the other two matrices, Drecent and Dresidual, as well.
We complemented our genetic studies with field research within the tribes by means of non-formal interviews with tribe members. Specifically, we interviewed old members of the sporadic tribes mentioned in this manuscript regarding tribe origin, dates of arrival to the Negev and their patterns of tribal intermarriages. All tribe members interviewed for this study claimed their origin from the Arabian Peninsular region hundreds of years before they migrated to the Middle East.
Results
We analyzed two data sets of Negev Bedouins: one from the HDGP (Jakobsson et al., 2008) and another set collected in our laboratory (LAB) having both related and unrelated individuals (Materials and methods and Supplementary Table S1). We defined recent, combined and residual ancestries as follows (See Supplementary text for detailed definitions). Recent ancestry refers to a common genetic ancestry from the most recent ∼1–20 generations. Combined ancestry refers to the last ∼1–50 generations. Residual ancestry refers to a common ancestry corresponding to ∼20–50 generations ago. These thresholds were chosen to reflect the average time of migration of the sporadic tribes and are based on a threshold for the length of shared segments. We defined the dissimilarity matrix to be the kinship matrix in logarithmic scale: D=−Log(kinship), and calculated three different dissimilarity matrices for each data set: Drecent, Dresidual and Dcombined. Intuitively, tribes that admixed only recently and do not have common clan ancestry are expected to show a signal in the recent dissimilarity matrix but not in the residual matrix. Tribes from the same clan are expected to show signals in all matrices, as they share a common history as well as interact in the present time.
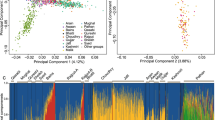
Previous work, analyzing the HDGP sample, already suggests that a fine structure within the Bedouin community is present (Lawson et al., 2012; Supplementary Figure S22). We start by focusing our analysis on the publicly available HDGP Bedouin data and further investigate the suggested patterns. Figure 1 presents the identification of the large clans in the HDGP data. Figure 1-a1 shows the distribution of the pairwise dissimilarity values in Dcombined. The distribution appears to be bimodal, which suggests that a structure is present. We intuitively hypothesized that the right mode (high D values) corresponds to the background sharing of unrelated pairs in the population, and the left mode (low D values) to the relatedness within tribes. Similar bimodal distributions were found for the other matrices Drecent and Dresidual as depicted in Supplementary Figure S1. A Gaussian Mixture Model (GMM) was fitted to generate the curved lines outlining the two populations (Supplementary Table S5). The intersection between the fitted Gaussians is indicated by a black dot. This is the location for which a naive Bayesian classifier would generate equal likelihoods for the two populations. In a Bayesian classifier, a pair of individuals with D below this point would be classified as related and above this point as unrelated.

The dissimilarity matrix for the HGDP data. (A) Panels a1–a3 depict the distribution of the pairwise dissimilarity values calculated for Dcombined, together with a fitted Gaussian Mixture Model. a2 emphasizes in gray shading the distribution of dissimilarity values within the clusters. a3 emphasizes in gray shading the dissimilarity values outside the clusters (the background distribution). B shows the permuted dissimilarity matrix. The color map for this figure is also depicted under the histograms, indicating that blue colors relate to the left mode and yellow to the right mode. The identified clusters are marked in B by a red line as b1, b2 and b3.
Figure 1B shows the permuted dissimilarity matrix Dcombined for the HDGP data. Each pair of individuals is represented by a colored rectangle. We used the GMM parameters to generate the color map by allowing blue colors to span the entire domain of the related distribution (the left mode) followed by a gradual hue change to green and yellow for the background distribution (the right mode). Figure 1B clearly shows one big cluster and two smaller ones. In order to show how these clusters relate to the bimodal distribution, we examined the distribution of dissimilarities within and between the clusters. We defined the borders of the three clusters manually (marked with red line), labeling them as b1, b2 and b3 (Figure 1B). The pairwise dissimilarity histograms within the clusters are emphasized in Figure 1-a2 and between the clusters in Figure 1-a3, clearly indicating that the left mode corresponds to relatives within clusters, whereas the right mode corresponds to unrelated pairs. Figure 1-a2 also suggests that distant relatives could reside well within the background distribution (right mode). A Bayesian classifier based on the bimodal distribution would mark all individuals with D>1.5 as unrelated. However, by analyzing all pairs simultaneously and identifying the clans, the relationship between seemingly unrelated pairs is established indirectly via relatedness with other members in the group. Thus, our analysis is not only effective in the delineation of clans but also enables the identification of very distant relatives with negligible genomic sharing simply because of common clan ancestry.
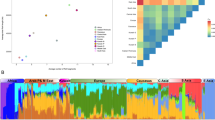
The HDGP data contain no genealogical knowledge of the individuals in the sample and therefore limits our ability to further study relationships between the tribes. We thus turn to the LAB data, in order to correlate the results with known genealogical information. In particular, we wanted to test the feasibility of differentiating between the large clans and the sporadic tribes that migrated sporadically to the Negev. In Figure 2, A and B depict the distribution of the pairwise dissimilarity values and the permuted matrix constructed from Dcombined. The known relatives are emphasized in the histogram with a typical D<1 value. The existence of a bimodal distribution is clear in this figure as well. Figure 2C shows the matrices for recent and residual ancestries with a few identified clusters depicted on each matrix. All matrices are shown using the color map generated from the HDGP data.

The dissimilarity matrix for the LAB data. a and b depict the distribution of the pairwise dissimilarity values and the permuted matrix constructed from Dcombined. The known relatives are emphasized in the histogram and have typical D<1 values. c shows the matrices for recent and residual ancestries, with a few identified clusters depicted on each matrix. All matrices are shown using the color map generated from the HDGP data. The index numbers in the matrices refer to individuals as depicted in Supplementary Table S1.
Two main clusters are obvious and are indicated in Figure 2 as A and B. It is evident from Figure 2C that both recent and ancient ancestries contribute to the signals in clusters A and B, as both clusters are clearly indicated in Drecent and Dresidual. Note, however, that cluster A seems to be distinct from the rest of the data, whereas cluster B shows signals of common ancestry with several external tribes around it. This signal is mostly absent from the residual matrix, suggesting recent tribal admixture. These patterns are more easily depicted in Figure 3, which is a binary version of the matrices in Figure 2. We generated Figure 3 using the Bayesian intersection points as thresholds for each matrix. Investigation of surnames identified cluster A as one of the oldest, well established clans in the Negev. On the other hand, cluster B is composed of related tribes, probably from a common founder, that migrated from Gaza to the Negev around 300 years ago. Thus, it seems that clan B, as opposed to clan A, allows interactions with tribes outside the clan. In fact, surname investigation suggests that, apart from clan A, all other tribes in our data are seemingly sporadic.

The recent and residual dissimilarity matrices from the LAB data with binary thresholds. We generated these matrices using the Bayesian intersection points as thresholds for each matrix. Dissimilarities below the threshold were colored black, whereas those above remained in white. Signals of recent ancestry that are not replicated in the residual ancestry matrix indicate recent admixture. Specifically, individuals 29 and 14 show substantial recent ancestry with clan B (3, 11, 24, 1, 8) but no residual ancestry.
More examples for the interplay between residual and recent ancestries are manifested in clusters D and C. There is a clear recent ancestry signal between the two tribes in cluster C that seems to be absent from the residual ancestry matrix. Consistent with this finding, our genealogical investigation revealed that these two tribes have coexisted side by side for the last ∼200 years; however, there is no record for common clan ancestry, as one tribe migrated from Egypt and the other from Jordan. On the other hand, cluster D, which appears in the residual ancestry matrix, is only partially depicted in the recent ancestry matrix overlapping the surrounding clusters C and B. We think that cluster D represents a noise artifact. We do not know of any common ancestry between the tribes of this cluster. Using the individual index numbers indicated in Figure 2, tribes represented by individuals 13 and 14 migrated from two different regions in Gaza; tribe represented by 17 is associated with Egyptian ancestry; and tribe represented by 7 migrated from Jordan. As explained in the appendices (Supplementary Text), the interpretation of the residual ancestry matrix in this case is problematic. We do not infer ancient ancestry directly, and as this cluster is not clearly indicated in the recent ancestry matrix, it is most likely a false-positive cluster. Indeed, cluster D is not manifested in the binary version of Figure 3 and does not survive repeated permutations on the residual matrix using SPIN.
In order to relate our findings to patterns of rare variants prevalent in the different tribes, we listed in Table 1 several founder mutations known to be associated with Mendelian disorders in the Bedouin community. For example, founder mutations for Hereditary Retardation Dysmorphism syndrome and for Congenital Insensitivity to Pain and Anhydrosis are known to be present in many tribes associated with clan A. In fact, the same founder mutation that causes Hereditary Retardation Dysmorphism in the Negev Bedouins was also found in cases from Saudi Arabia (Teebi, 1997). This fact triggered a global screening for the founder Hereditary Retardation Dysmorphism mutation in Bedouins from all tribes appealing for genetic counseling. However, at least for the tribes participating in this study, we find no evidence for the founder mutation outside of clan A (see Table 2). As a second example, two other founder mutations, Mitochondrial Complex 3 Deficiency (Complex 3) and Persistent Hyperinsulinemic Hypoglycemia of Infancy, are found exclusively in clan B. However, at least one founder mutation associated with deafness is found both in clan B and in one of the sporadic tribes. As we see significant recent kinship between clan B and other sporadic tribes, we expect to detect more such common mutations in the near future.
Discussion
Our results indicate that we can identify Bedouin clans as large extended families in which each individual is related to many others in the group. We showed that clans encompass both recent and ancient common ancestry as expected. Furthermore, our analysis identifies tribes that were ‘allowed’ to intermarry versus those whose mixture was socially forbidden, thus unraveling relationships consistent with the known historical background of the Negev Bedouins.
Previous studies of Bedouin tribes were focused mainly on the relationships between the Bedouins with their surrounding populations. For example, Abu-Amero et al. (2008, 2009) used non-recombinant Y and mitochondrial DNA from Saudi Arab samples for the study of migration patterns from Africa to Asia in ancient times. Their work was focused on large-scale migration patterns, such as the introduction of African haplotypes into Arabian Peninsular populations. Mohammad et al. (2009) studied a more recent history of population structure in Kuwait. They used both autosomal and Y chromosomal markers to study samples from six Kuwaiti tribes. They found a remarkable correspondence between tribe names and their genetic makeup, supporting the expected traditional history and tribal isolation. More recently, Hunter-Zinck et al. (2010) studied the admixture patterns in the population of Qatar and recovered three affinity groups within the population consistent with the migration patterns into the area.
The studies mentioned above recovered much of the expected Bedouin population structure using methods that associate haplotype frequencies with ancestry. Utilizing haplotype frequencies to study structure is suitable when a population exhibits dramatic migration or admixture patterns with the surrounding populations, so that certain haplotype frequencies would differ significantly between different groups. In our study of the Negev Bedouins, we took a complementary approach that allowed us to look at more subtle signals of population structure. As our sample is entirely from a recent Arabian origin, classical admixture analysis using principle component analysis or Bayesian models might not be adequate (data not shown) (Falush et al., 2003; Patterson et al., 2006). Our prior knowledge regarding the Bedouin history and the segregation patterns of rare diseases in the different tribes suggests the analogy between tribes and extended families and thus leads naturally to IBD analysis. Thus, IBD analysis seems to compliment these other studies by revealing a fine structure within a seemingly homogeneous sample of Bedouins tribes.
We note that the knowledge gained in our study is extremely important for IBD mapping in sporadic cases. Recently, we mapped a new gene for lethal congenital contracture syndrome type 4 (LCCS4) (Markus et al., 2012) in tribes N and O (Table 1; represented by individuals 13 and 14 in Figure 2). Consistent with the lack of significant recent ancestry or clan co-ancestry, the shared locus resides in a very small segment, roughly 1 Mbp in size, indicating relatively ancient ancestry. Two other sporadic cases that manifest the LCCS phenotype and do not posses any of the known mutations were also analyzed using our methodology. These two cases were found to share common ancestry with clan B (data not shown). We have thus initiated a search for yet another LCCS founder mutation expected to be present in cases from clan B.
Several drawbacks of this approach should be indicated. First, we did not try to cluster the samples into subgroups. Clustering ultimately involves loss of information, whereas the visual inspection of the sorted matrices still leaves much room for interpretation. Eventually, there is a need to make decisions regarding the significance of the kinship signals. Currently, we use a conservative approach when testing for relatedness. As indicated above, we use the Bayesian classifier over the distribution of dissimilarity values to assess the significance of pairwise relatedness and refer to the sorted dissimilarity matrices as a visual compliment. More work is needed to facilitate the knowledge embedded in the sorted matrices for the assessment of relatedness beyond pairwise kinship scores.
Secondly, there is a limit to the resolution of this approach. Although the ancestry thresholds used here seem to work well for the clustering of Bedouin tribes, they might not suit the analysis of fine structure in other populations. Recent studies on models of migration suggest that the distribution of the lengths of shared genomic segments could be used to infer ancestry more accurately (Huff et al., 2011; Pool and Nielsen, 2009; Henn et al., 2012). Adaptation of these methods for fine structure analysis in small isolated populations should give a more accurate basis for both the ancestry inference and the clustering challenges (Novembre and Ramachandran, 2011).
Despite these caveats, the emergent structure is well correlated with patterns of rare variants known to us. The recent technology advances and the growing awareness for genetic disorders in the Bedouin community are having a large impact on the identification of deleterious mutations in the Bedouin population. Currently, there are >40 different mutations known to us, most are tribe-specific. In genetic counseling to the Bedouin community, we offer members of different tribes’ carrier testing for different mutations, based on their tribal prevalence. The current study, delineating genomic similarities between tribes, enables us to better identify tribes sharing identical mutations, improving our ability to suggest appropriate carrier testing. Thus, our findings here are conducive to a more detailed and adequate carrier-screening program for rare disorders shared by various tribes, with the purpose of serving this community more effectively.
Data archiving
Data deposited in the Dryad repository: doi:10.5061/dryad.1s2j3.
References
Abecasis GR, Cherny SS, Cookson WO, Cardon LR . (2001). Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30: 97–101.
Abu-Amero KK, Hellani A, González AM, Larruga JM, Cabrera VM, Underhill PA . (2009). Saudi Arabian Y-Chromosome diversity and its relationship with nearby regions. BMC Genet 10: 59.
Abu-Amero KK, Larruga JM, Cabrera VM, González AM . (2008). Mitochondrial DNA structure in the Arabian Peninsula. BMC Evol Biol 8: 45.
Abu-Rabia A . (1994). The Bedouin Refugees in the Negev. Refuge: Canada's periodical on refugees 14: 15–17.
Al-Aref A . (1934). The Bedouin tribes in Beer-Sheva district. Bostnai: Tel Aviv.
Astle W, Balding DJ . (2009). Population structure and cryptic relatedness in genetic association studies. Stat Sci 24: 451–471.
Atzmon G, Hao L, Pe'er I, Velez C, Pearlman A, Palamara PF et al. (2010). Abraham's children in the genome era: major Jewish diaspora populations comprise distinct genetic clusters with shared Middle Eastern Ancestry. Am J Hum Genet 86: 850–859.
Bailey C . (1985). Dating the arrival of the Bedouin Tribes in Sinai and the Negev. J Econ Soc Hist Orie 20–49.
Barel O, Shorer Z, Flusser H, Ofir R, Narkis G, Finer G et al. (2008). Mitochondrial complex III deficiency associated with a homozygous mutation in UQCRQ. Am J Hum Genet 82: 1211–1216.
Bercovici S, Meek C, Wexler Y, Geiger D . (2010). Estimating genome-wide IBD sharing from SNP data via an efficient hidden Markov model of LD with application to gene mapping. Bioinformatics 26: 175–182.
Browning SR . (2008). Estimation of pairwise identity by descent from dense genetic marker data in a population sample of haplotypes. Genetics 178: 2123–2132.
Cavalli-Sforza LL, Menozzi P, Piazza A . (1994) The History and Geography of Human Genes. Princeton university press: Princeton, New Jersey, USA.
Chatty D . (2006) Nomadic Societies in the Middle East and North Africa: Entering the 21st Century. Brill Academic Pub: Leiden, The Netherlands.
Falush D, Stephens M, Pritchard JK . (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164: 1567–1587.
Finer G, Shalev H, Birk OS, Galron D, Jeck N, Sinai-Treiman L et al. (2003). Transient neonatal hyperkalemia in the antenatal (ROMK defective) Bartter syndrome. J Pediatr 142: 318–323.
Gasbarra D, Pirinen M, Sillanpää MJ, Salmela E, Arjas E . (2007). Estimating genealogies from unlinked marker data: a Bayesian approach. Theor Popul Biol 72: 305–322.
Glaser B, Phillip M, Carmi R, Lieberman E, Landau H . (1990). Persistent hyperinsulinemic hypoglycemia of infancy (‘nesidioblastosis’): autosomal recessive inheritance in 7 pedigrees. Am J Med Genet 37: 511–515.
Gusev A, Lowe JK, Stoffel M, Daly MJ, Altshuler D, Breslow JL et al. (2009). Whole population, genome-wide mapping of hidden relatedness. Genome Res 19: 318–326.
Henn BM, Botigué LR, Gravel S, Wang W, Brisbin A, Byrnes JK et al. (2012). Genomic ancestry of North Africans supports back-to-Africa migrations. PLoS Genet 8: e1002397.
Huff CD, Witherspoon DJ, Simonson TS, Xing J, Watkins WS, Zhang Y et al. (2011). Maximum-likelihood estimation of recent shared ancestry (ERSA). Genome Res 21: 768–774.
Hunter-Zinck H, Musharoff S, Salit J, Al-Ali KA, Chouchane L, Gohar A et al. (2010). Population genetic structure of the people of Qatar. Am J Hum Genet 87: 17–25.
Jakobsson M, Scholz SW, Scheet P, Gibbs JR, VanLiere JM, Fung HC et al. (2008). Genotype, haplotype and copy-number variation in worldwide human populations. Nature 451: 998–1003.
Kirkpatrick B, Li S, Karp R, Halperin E . (2011). Pedigree reconstruction using identity by descent. RECOMB 6577: 136–152.
Laberge AM, Michaud J, Richter A, Lemyre E, Lambert M, Brais B et al. (2005). Population history and its impact on medical genetics in Quebec. Clin Genet 68: 287–301.
Lander ES, Green P . (1987). Construction of multilocus genetic linkage maps in humans. Proc Natl Acad Sci USA 84: 2363–2367.
Lawson DJ, Hellenthal G, Myers S, Falush D . (2012). Inference of population structure using dense haplotype data. PLoS Genet 8: e1002453.
Leal SM, Yan K, Müller-Myhsok B . (2005). SimPed: a simulation program to generate haplotype and genotype data for pedigree structures. Hum Hered 60: 119–122.
Markus B, Birk OS, Geiger D . (2011). Integration of SNP genotyping confidence scores in IBD inference. Bioinformatics 27: 2880–2887.
Markus B, Narkis G, Landau D, Birk RZ, Cohen I, Birk OS . (2012). Autosomal recessive lethal congenital contractural syndrome (LCCS4) caused by a mutation in MYBPC1. Hum Mutat 33: 1435–1438.
Marx E . (1967) Bedouin of the Negev. Manchester University Press: Manchester, England.
Marx E . (1977). The tribe as a unit of subsistence: nomadic pastoralism in the Middle East. Am Anthropologist 79: 343–363.
Mathieson I, McVean G . (2012). Differential confounding of rare and common variants in spatially structured populations. Nat Genet 44: 243–246.
Mohammad T, Xue Y, Evison M, Tyler-Smith C . (2009). Genetic structure of nomadic Bedouin from Kuwait. Heredity 103: 425–433.
Narkis G, Landau D, Manor E, Elbedour K, Tzemach A, Fishelson M et al. (2004). Homozygosity mapping of lethal congenital contractural syndrome type 2 (LCCS2) to a 6 cM interval on chromosome 12q13. Am J Med Genet 130: 272–276.
Novembre J, Ramachandran S . (2011). Perspectives on human population structure at the Cusp of the sequencing era. Annu Rev Genomics Hum Genet 12: 245–274.
Parvari R, Hershkovitz E, Grossman N, Gorodischer R, Loeys B, Zecic A et al. (2002). Mutation of TBCE causes hypoparathyroidism–retardation–dysmorphism and autosomal recessive Kenny–Caffey syndrome. Nat Genet 32: 448–452.
Patterson N, Price AL, Reich D . (2006). Population structure and eigenanalysis. PLoS Genet 2: e190.
Peltonen L, Palotie A, Lange K . (2000). Use of population isolates for mapping complex traits. Nat Rev Genet 1: 182–190.
Pool JE, Hellmann I, Jensen JD, Nielsen R . (2010). Population genetic inference from genomic sequence variation. Genome Res 20: 291–300.
Pool JE, Nielsen R . (2009). Inference of historical changes in migration rate from the lengths of migrant tracts. Genetics 181: 711–719.
Price AL, Zaitlen NA, Reich D, Patterson N . (2010). New approaches to population stratification in genome-wide association studies. Nat Rev Genet 11: 459–463.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575.
Scott DA, Carmi R, Elbedour K, Duyk GM, Stone EM, Sheffield VC . (1995). Nonsyndromic autosomal recessive deafness is linked to the DFNB1 locus in a large inbred Bedouin family from Israel. Am J Hum Genet 57: 965.
Sheffield VC, Stone EM, Carmi R . (1998). Use of isolated inbred human populations for identification of disease genes. Trends Genet 14: 391–396.
Strauss KA, Puffenberger EG . (2009). Genetics, medicine, and the plain people. Annu Rev Genomics Hum Genet 10: 513–536.
Sun L, Wilder K, McPeek MS . (2002). Enhanced pedigree error detection. Hum Hered 54: 99–110.
Teebi AS . (1997) Genetic Disorders Among Arab Populations. Oxford University Press: New York, NY, USA.
Tsafrir D, Tsafrir I, Ein-Dor L, Zuk O, Notterman D, Domany E . (2005). Sorting points into neighborhoods (SPIN): data analysis and visualization by ordering distance matrices. Bioinformatics 21: 2301.
Weir BS, Anderson AD, Hepler AB . (2006). Genetic relatedness analysis: modern data and new challenges. Nat Rev Genet 7: 771–780.
Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF et al. (2005). A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38: 203–208.
Acknowledgements
We thank Dan Geiger, Eytan Domany, Amit Zeisel and Noa Bossel for their helpful discussions and Assif Yitzhaky for sharing the SPIN code. This study was supported by the Legacy Heritage Bio-Medical Program of the Israel Science Foundation (grant no. 1520/09) and through the Kahn Family Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Markus, B., Alshafee, I. & Birk, O. Deciphering the fine-structure of tribal admixture in the Bedouin population using genomic data. Heredity 112, 182–189 (2014). https://doi.org/10.1038/hdy.2013.90
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2013.90
