
Abstract
The mapping of sterile genes is an essential issue, which should be solved for the investigation of sterility mechanism in wide hybridization of plants. However, the methods formerly developed cannot address the problem of mapping sterile loci with epistasis. In this study, we developed a new method to map sterile genes with epistasis in wide hybridizations of plants using a backcross design. The maximum likelihood method was used to estimate the parameters of recombination fractions and effects of sterile genes, and the convergent results of these parameters were obtained using the expectation maximization (EM) algorithm. The application and efficiency of this method were tested and demonstrated by a set of simulated data and real data analysis. Results from the simulation experiments showed that the method works well for simultaneously estimating the positions and effects of sterile genes, as well as the epistasis between sterile genes. A real data set of a backcross (BC) population from an interspecific hybrid between cultivated rice, Oryza sativa, and its wild African relative, Oryza longistaminata, was analyzed using the new method. Five sterile genes were detected on the chromosomes of 1, 3, 6, 8 and 10, and significant epistatic effects were found among the four pairs of sterile genes.
Similar content being viewed by others


Introduction
Wide hybridization is a useful tool in plant breeding. It provides us with a means of transferring the desirable characters from wild species into cultivated species, and can be used to introduce alien variation, study genome structure and phylogeny, and sometimes may be used to produce a new species or a new crop. However, crossing plants from two different species is more challenging and has limited success. There are three main barriers to wide hybridization: (i) incompatibility between parent species; (ii) inviability of the F1 hybrid; and (iii) sterility of the F1 hybrid or its progeny (Sharma and Gill, 1983). Some of these are prezygotic barriers that occur before fertilization, whereas some are postzygotic barriers that occur after fertilization. Postzygotic barriers may lead to the isolation of different populations by reducing hybrid fitness (López-Fernández and Bolnick, 2007). Hybrid sterility is one of the postzygotic barriers, in which the hybrids that survive are unable to reproduce their offspring. Such reproductive barriers generally hinder transferring of genes for useful characters in the cross-breeding of crops. Despite the importance of these barriers, little is made clear about the kind of molecules related to hybrid sterility and how they arise in the course of speciation (Kubo et al., 2008). Thus, to understand the genetic mechanism of postzygotic barriers in wide hybridization of plants, we must identify the genes affecting fertility.
The location and effect of reproductive barriers have been estimated historically by analyzing their association with a mapped morphological trait or biochemical trait loci. However, high-resolution mapping has clearly revealed the genetic architectures underlying the sterility of hybrids between different species. Genetic linkage maps are useful tools for mapping traits of interest and for studying the evolution of genomic organization (Fishman and Willis, 2001; Chen and Tanksley, 2004). For instance, interspecific linkage mapping makes it possible to locate the genetic basis of reproductive isolating mechanisms in natural populations that may lead to speciation (Moyle and Nakazato, 2008) and genetic regions that control mating system variation (Chen and Tanksley, 2004; Chen et al., 2007).
DNA markers can be used to analyze quantitative trait loci (QTL) that may contribute to reproductive isolation. However, four important limitations exist in the study of reproductive barriers using QTL analysis. First, as a phenotype of an individual, sterility is determined both by genotypes of its own and by genotypes of its progeny. Without information of the genotypes of the aborted gametes and zygotes, it would be difficult to use the QTL method to analyze hybrid sterility. Second, the traits selected to be investigated are restricted to those believed, a priori, to be involved in the isolation mechanisms. Third, it is difficult to compare the isolation efficiency among different traits detected as reproductive barriers by QTL analysis. Finally, deviations in allele frequencies can weaken the statistical sensitivity for the identification of QTL, which could affect sterility or inviability at the reproductive barrier (Harushima et al., 2001). DNA markers can also be used to study reproductive barriers by analyzing deviations from expected Mendelian segregation ratios. Hybrid sterility genes can lead to segregation distortions at such loci and at the marker loci linked to them. Several studies have been reported for investigating reproductive barriers by the analysis of segregation distortions of DNA markers. Cheng et al. (1996) presented a method for estimating the recombination fractions between a lethal factor locus and molecular markers nearby; the relative viability of gametes or zygotes affected by the lethal factor was obtained using the maximum likelihood (ML) approach and the expectation conditional maximization (ECM) algorithm in an F2 population. The ML method, the bayesian method and the multiresponse nonlinear regression method based on molecular markers were developed to map segregation-distorting loci (SDL) that caused deviations from expected Mendelian segregation ratios in different crops (Vogl and Xu, 2000; Harushima et al., 2001; Harushima et al., 2002; Lu et al., 2002). Luo and Xu (2003) extended the ML method to map viability loci in outbred populations, which used the observed marker genotypes as data and the proportions of the genotypes of the viability locus as parameters. In two F2 populations of rice (Oryza sativa L.), a multipoint method of ML was used to estimate the positions and effects of the SDLs (Wang et al., 2005). The agreement between chromosomal regions exhibiting marker transmission ratio distortion (TRD) and QTLs known to contain hybrid incompatibility was evaluated qualitatively and quantitatively in a genome-wide scale (Moyle and Graham, 2006). A multipoint ML method was proposed for estimating the location and the effect of SDL under the liability model along with selection coefficients of marker genotypes and recombination fractions (Zhu and Zhang, 2007). Chen and Walsh (2009) presented an ML method for genetic mapping of a female partial-sterile locus in a backcross population using the genotype data of molecular markers. However, a common limitation in the methods mentioned above is that they cannot address the problem of mapping sterile loci with epistasis.
Dobzhansky (1936) and Muller (1942) suggested a two-locus model in which deleterious epistatic interaction among different loci results in hybrid inviability. This model is still widely accepted in plants and animals, and can also be used for the analysis of hybrid sterility. In addition, hybrid sterility shows a typical polygenic inheritance; hence, it is conceivable that the interaction among the polygenes is related to the sterility mechanism (Kubo et al., 2008). Recent studies have mapped the epistatic genes underlying hybrid sterility and inviability in diverse animal species such as Drosophila (Orr and Irving, 2001; Chang and Noor, 2010) and in plants such as Solanum (Moyle and Nakazato, 2009) and rice (Kubo et al., 2008; Garavito et al., 2010; Kubo et al., 2011). Epistasis in more than two loci could therefore be fundamentally important in determining patterns and rates of evolution of isolation between diverging species. Nonetheless, the prevalence and nature of interactions between more than two loci from one or both species involved in a hybridization (that is , ‘complex epistasis’), and their effects on hybrid incompatibility, are poorly understood empirically (Moyle and Nakazato, 2009).
In this study, we developed a new method for mapping sterile genes with epistasis in wide hybridizations of plants using a BC design. The ML method was used to estimate the location and intensity of the reproductive barriers. The relative viability of a gamete carrying a sterile allele was also estimated as compared with that of a normal one. The ML solutions of the parameters were obtained via the expectation maximization (EM) algorithm (Dempster et al., 1977). To illustrate the method, a simulation study was presented. As an application of the proposed method, the reproductive barriers of an interspecific hybrid between cultivated rice, O. sativa, and its wild African relative, Oryza longistaminata, were analyzed, using a saturated molecular map (Causse et al., 1994).
Theory
Genetic model
We developed the model under a BC design. The parents that initiate the cross were assumed to be pure inbred lines. The parents P1 (m1m1s1s1m2m2, m3m3s2s2m4m4) and P2 (M1M1S1S1M2M2, M3M3S2S2M4M4) were crossed to produce the F1 hybrid, where M1-m1 and M2-m2 are two molecular markers flanking the sterile locus S1-s1, M3-m3 and M4-m4 are two molecular markers flanking the sterile locus S2-s2, and S1-s1 and S2-s2 are two non-linked loci that are located on different chromosomes. The order of the loci is M1-S1-M2 and M3-S2-M4. The recombination values between M1 and S1, S1-M2, M3-S2 and S2-M4 are r1, r2, r3 and r4, respectively. The F1 hybrid from P1 × P2 cross is backcrossed to one of its parents (here P2) to produce a BC population of n individuals. When the F1 of the cross is used as the maternal parent, the female sterility loci can be mapped, and when the F1 of the cross is used as the paternal parent the male sterility loci can be mapped. We only consider the situation in which the F1 of the cross is used as the maternal parent (F1 × P2), in which female gametes generated by the F1 hybrid are subjected to the selection of sterile genes s1 and s2. The viability of female gametes with haplotype s1S2, S1s2 and s1s2, relative to the normal gametes with haplotype S1S2, are u, v and x, respectively, where x denotes the epistasis between sterile genes s1 and s2.
We aim to map loci that may contribute to female gamete sterility using mapped molecular markers on the genome. The parameters are the positions, effects and epistatic effects of these loci. For the six loci in the model, there are 64 kinds of offspring from the F1 hybrid in the BC population. However, we cannot observe the genotypes of sterile loci S1-s1 and S2-s2 directly, and the genotypic data here are the observed marker genotypes only. The expected frequencies of the 16 kinds of genotypes at M1-M2 and M3-M4 in the BC population are listed in Table 1.
Parameter estimates
We set n as the total number of individuals in the BC population, and ni (i=1, 2, …, 16) as the observed phenotypic counts of the 16 kinds of genotypes at marker loci M1, M2, M3 and M4. Then the likelihood function is as follows:

where pi (i=1,2,…16) is the expected frequency of the ith phenotypic class at molecular marker loci M1, M2, M3 and M4. The data are the phenotypic counts corresponding to the molecular marker genotypes listed in Table 1. The parameters including recombination fractions r1, r2, r3, r4, and the differential viability u, v and x are estimated in this study.
Theoretically, the Newtow–Raphson method can be used to obtain the ML estimates of parameters in equation (1), but the second derivatives are complex, and it is very difficult to obtain the explicit solutions for all parameters. Hence, the EM algorithm (Dempster et al., 1977) was applied to estimate the parameters.
We assumed that the four kinds of genotypes S1S2, s1S2, S1s2 and s1s2 are observable in the complete data set, although they are actually missing data. When all the 64 genotypes of markers and sterile loci were observable, the complete information likelihood function could be expressed as follows:

where pij and nij are the expected frequency and the observed number of the jth sterile genotype in the ith marker genotype, respectively. The partial derivative of the kth parameter in the logarithm likelihood is as follows:

where θk=r1,r2,r3,r4,u,v,x.
Setting the above partial derivatives to zero and then solving the equations, we can get the ML estimates. The iterative expressions of the parameters are as follows:







As the nij values in the above equations are not the observed genotype counts, the parameters cannot be estimated directly. However, the EM algorithm could solve this issue, which includes the following steps:
(1) Initializing the parameters: the initial value of recombination fraction rk(0)(k=1, 2, 3, 4) is one of the numbers between 0 and 0.5, and the viability coefficient (u, v, x) is initialized with 1;
(2) The E-step is to calculate the counts nij by using pij, pi and genotype counts ni:

(3) The M-step is to update the parameter estimates by equations (4, 5, 6, 7, 8, 9, 10).
Iterate the E-step and the M-step until convergence is reached, that is, |θ(k+1)−θ(k)|<10−6.
Significance test
The null hypothesis H0: u=1, v=1, x=1 was applied to test the sterile loci without viability selection, and the statistic of the likelihood ratio (LR) test can be expressed as follows:

where L(·) is the logarithm likelihood, and LR1 approximately follows the χ2 distribution with three degrees of freedom ( ) under the H0 hypothesis. Furthermore, to test whether the epistatic effects exist, another null hypothesis H0: x=1 was adopted. The LR statistic is:
) under the H0 hypothesis. Furthermore, to test whether the epistatic effects exist, another null hypothesis H0: x=1 was adopted. The LR statistic is:

LR2 approximately follows a  distribution. To control the false discovery rate (FDR), we took the multiple test proposed by Bonferroni, with α=α*/N, where N is the number of hypotheses and α* is the value of significance level 0.01 or 0.05. The set of tests to which this procedure is applied consists of all the null hypotheses corresponding to the combinations of marker intervals involved in the genome scan for the sterile genes.
distribution. To control the false discovery rate (FDR), we took the multiple test proposed by Bonferroni, with α=α*/N, where N is the number of hypotheses and α* is the value of significance level 0.01 or 0.05. The set of tests to which this procedure is applied consists of all the null hypotheses corresponding to the combinations of marker intervals involved in the genome scan for the sterile genes.
Applications
To illustrate the application of this method, simulation experiments were conducted, and the real data set from one cross in rice was analyzed, which was generated between cultivated species of rice (O. sativa) and its wild African relative (O. longistaminata).
Monte Carlo simulation
In the simulation experiments, two chromosomes were simulated. One of them was spaced equally with 10 markers, the total length of which was 180 cM; the other contained eight markers equally distributed and the total length was 140 cM. Two sterile loci s1 and s2 were located in the two chromosomes, s1 was in the interval between marker six (denoted as 1M6) and marker seven (denoted as 1M7) on the first chromosome, s2 was in the interval between marker four (denoted as 2M4) and marker five (denoted as 2M5) on the second chromosome. The genetic distance between s1 and 1M6 was 9 cM and that between s1 and 1M7 was 11 cM. The genetic distance between s2 and 2M4 was 12 cM and that between s2 and 2M5 was 8 cM. Three factors considered in these simulation experiments were the sample size of the population (ranging from 50 –500), the intensity of viability (0–1) and the recombination fraction between sterile loci and linked markers (0–0.5). Each process was carried out with 1000 replicates, and the performances of all parameter combinations were evaluated using the bias and standard deviations of these replicates.
A data set with 300 individuals was simulated as an example and the estimates of parameters are listed in Table 2. In this example, only in the intervals where the putative sterile genes were located, that is, the interval between markers 1M6 and 1M7 (designated as 1M67) and the interval between 2M4 and 2M5 (designated as 2M45), were the estimates for the seven parameters very close to their true values. The farther the interval markers are from the flanking marker combination 1M67-2M45, the greater the estimates of viability (u, v and x) tend to deviate from the respective true values. When the markers were farther enough from the sterile loci, the estimates of viability (u, v and x) tend to be 1, which means that these markers are in normal segregation.
The LR statistics for the tests on the presence of viability selection (LR1) and epistasis between loci (LR2) are obtained using the estimates of the seven parameters. The results are shown in Figure 1. The maximum values of LR1 and LR2 are both observed at the sixth interval marker (1M67) and the fourth interval (2M45) of the two chromosomes, that is, the flanking marker combination 1M67-2M45, which contains the sterile genes s1 and s2. The cutoff α values in the multiple tests for LR1 are 7.94e−4 (α*=0.05) and 1.59e−4 (α*=0.01), and those for LR2 are 9.40e−4 (α*=0.05) and 1.11e−4 (α*=0.01). The P-values of the χ2-test for the seven estimates in the 1M67-2M45 are very significant (P<1.59e−4). This example shows that the estimates obtained with the new method are unbiased and the hypothesis tests behave as expected.

The likelihood ratio (LR) value of the hypothesis test in the Monte Carlo simulation. (a) The test of viability selection; (b) the test of epistasis.
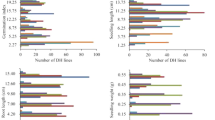
There are three factors that influence the estimation of parameters, including sample size for the mapping population, viability intensity and recombination fractions between the sterile genes and flanking markers. The effects of these factors on the estimation of parameters are presented in Figure 2, where the standard deviations and the bias from 1000 replicates are plotted. The results show that: (1) when the sample size of the BC population is larger than 100, the estimates for the locations and effects of the sterile genes are nearly unbiased and the increase in sample size can decrease bias and make it close to zero ultimately (Figure 2a); meanwhile, their standard deviations are also small and decrease with increasing sample size. (Figure 2b). (2) When the parameter r1 is less than 0.35, little bias is introduced in the estimates of other parameters; when the recombination fraction r1 is larger than 0.35, the bias of parameters u and r2 increases sharply. This may be due to the fact that the parameters u, r2 and r1 are all about the same sterile gene s1 in the same interval; simultaneously, the standard deviation has a similar trend with bias of parameters (Figure 2d). (3) As viability effect u increases from 0 to1, the bias and the standard deviation of the parameters r1, r2 and u increase gradually; however, all these values are smaller than 0.15, and other parameters are unaffected (Figures 2e and f). These results demonstrate that the proposed method is the most efficient when the sample size is large (>100), recombination is small, and there are strong viability effects.

The influence of sample size, recombination fraction and viability on the estimates of parameters. a and b show the bias and sd of r, u, v and x in different sample sizes (r1=0.15, r2=0.1, r3=0.12, r4=0.2, u=0.1, v=0.2, x=0.05); c and d show the bias and sd of r, u, v and x in different values of recombination fraction r1 (r2=0.1, r3=0.12, r4=0.2, u=0.1, v=0.2, x=0.05); e and f show the bias and sd of r, u, v and x in different values of viability u (r1=0.15, r2=0.1, r3=0.12, r4=0.2, v=0.2, x=0.1).
Real data analysis
As an application of the new method, a real data set of a rice BC population was analyzed using a saturated molecular map. This population (O. sativa/O. longistaminata//O. sativa) stemmed from an interspecific hybrid between two species of rice, cultivated (O. sativa) and its African wild relative (O. longistaminata) (Causse et al., 1994). The RFLP mapping data from the O. sativa × O. longistaminata cross were provided by Susan R. McCouch from the Department of Plant Breeding and Genetics, Cornell University. O. longistaminata species are isolated from the other species of AA genome by a strong reproductive barrier (Causse et al., 1994). The molecular map comprised 726 markers, and five sterile genes, named sb1, sb3, sb6, sb8 and sb10, were detected on the 1, 3, 6, 8 and 10 chromosomes. The locations and epistatic effects of these sterile loci are shown in Tables 3 and 4 respectively. We found significant epistasis in four pairs of sterile genes (Figure 3). As the population was generated by crossing the interspecific hybrid F1 as the female parent to the cultivated rice as the male parent, the sterile genes detected should be female sterile genes. The same sterile gene got similar estimates of viability effects in different marker combinations (Table 4). Table 4 shows that the effect of epistasis from sterile genes at least less than the effect of one sterile gene, and also exist the situation under which the epistasis effect less than two sterile genes effects.

The position and epistasis of sterile genes in the BC population (Oryza sativa/Oryza longistaminata//Oryza sativa). The broken lines indicate the location in the flanking markers, and double-arrow polylines connect the interacting loci and indicate that epistatic effects were detected among four pairs of sterile loci in the linkage map.
Discussion
Sterile genes in wide hybridizations have already been reported in several species in plants and animals. Genetic mapping of sterile genes plays a very important role in theoretical research and practical application (Song et al., 2005). The traditional methods for mapping sterile genes often depend on the data of seed set in the plant, rate of pollen stained with I2-KI, or embryo-sac morphology (Chen and Walsh, 2009, and references therein). These traits are vulnerable to environments and the criterion of sterility level is not consistent among different researchers. Some methods have been developed to map sterile genes or segregation distortion loci (Vogl and Xu, 2000; Luo and Xu, 2003; Luo et al., 2004; Wang et al., 2005; Chen and Walsh, 2009). These approaches take advantage of using the distorted segregating molecular markers linked to sterile genes, which circumvents the problems with seed set, rate of aborted pollen or embryo-sac morphology, but none of them took into account the existence of epistasis among sterile loci. Although epistasis is commonly believed to play a role in hybrid sterility, few studies have been conducted to detect the epistatic effects between sterile loci contributing to the unfitness of the hybrid. In this paper, we presented a statistical method using distorted segregating markers for mapping sterile genes with epistasis in wide hybridizations of plants in a BC population. The new method can effectively avoid the shortcoming of environmental influences and the inconsistency in the criteria of sterility.
In classical genetics the term epistasis refers to several kinds of gene interactions, including complementary, additive, duplicate effect etc. (Phillips, 1998). These effects cause the fixed segregation ratio of traits in the offspring population by which the type of epistasis can be defined. However, in the most practical situation, the epistasis type is difficult to distinguish completely because of the complex relation of genes and the influence of the environment. When the general traits are subject to multipoint gene interactions, the quantitative method is utilizable. However, the epistasis of sterile genes is different from traditional epistasis. The effect of some sterile genes can cause an individual to be dead, and when there are many sterile genes in an individual the expression in one sterile gene may affect that in others; thus, the epistatic interactions among them are complicated. For studying the epistasis of sterile genes, the epistasis considered in this paper is taken as a single parameter, which may include different kinds of epistasis in its conventional definition; this can solve the practical problem and lower the complexity of the method for estimating.
The epistatic effects have been found among four pairs of sterile genes in the real data analysis in the BC population from interspecific hybridization rice by our method. From the relation of epistasis effect (x) and the main effects of sterile genes (u, v), we find that the parameter x is less than one of the two main effects or less than both of them. It is different from the situation of dominant epistasis or recessive epistasis in the classical genetics, which is in accordance with the model and the assumptions we proposed completely. The parameter x is the epistasis effect of two sterile genes existing in a gamete; u and v are the effects of only one sterile gene. In the process of gamete generation, the gametes include 0, 1 and 2 sterile genes whose viability effect corresponds to 1, u or v, x, respectively. The result confirmed the feasibility of our method in practical application.
The method presented in this paper uses the flanking markers in a BC population. We also developed the method using the single marker and flanking markers in two different chromosomes, which is similar to the interval method except that one less solution of recombination fraction is to be estimated. The interval method makes use of the largest amount of marker information, and hence it is more accurate for parameter estimation; however, the calculating amount is great while scanning the whole genome. When one sterile gene in the chromosome is a candidate gene and linked closely with the marker, such as wide compatibility gene s5n (Ouyang et al., 2010), the loci-interval method is suggested to detect epistasis among s5n and other loci. When sterile genes are unknown the method in this paper is more suitable. For species in which it is easy to obtain a BC population, such as rice, our method is useful. However, for difficult cross-species, such as soybean, F2 rather than backcross population is more frequently used for mapping. Further studies are required to develop methods for other types of experimental designs.
Data Archiving
The RFLP mapping data analyzed in our study was provided by Susan R. McCouch from the Department of Plant Breeding and Genetics, Cornell University. The data can be accessed and downloaded from the Cornell University website at: http://ricelab.plbr.cornell.edu/Causse_at_al_1994.
References
Causse MA, Fulton TM, Cho YG, Ahn SN, Chunwongse J, Wu K et al. (1994). Saturated molecular map of the rice genome based on an interspecific backcross population. Genetics 138: 1251–1274.
Chang AS, Noor MAF . (2010). Epistasis modifies the dominance of loci causing hybrid male sterility in the drosophila pseudoobscura species group. Evolution 64: 253–260.
Chen J, Walsh B . (2009). Method for the mapping of a female partial-sterile locus on a molecular marker linkage map. Theor Appl Genet 119: 1085–1091.
Chen KY, Cong B, Wing R, Vrebalov J, Tanksley SD . (2007). Changes in regulation of a transcription factor lead to autogamy in cultivated tomatoes. Science 318: 643–645.
Chen KY, Tanksley SD . (2004). High-Resolution Mapping and Functional Analysis of se2.1. Genetics 168: 1563–1573.
Cheng R, Saito A, Takano Y, Ukai Y . (1996). Estimation of the position and effect of a lethal factor locus on a molecular marker linkage map. Theor Appl Genet 93: 494–502.
Dempster AP, Laird NM, Rubin DB . (1977). Maximum likelihood from incomplete data via the em algorithm. J Roy Stat Soc. B (Met) 39: 1–38.
Dobzhansky T . (1936). Studies on hybrid sterility. II. Localization of sterility factors in Drosophila pseudoobscura hybrids. Genetics 21: 113–135.
Fishman L, Willis JH . (2001). Evidence for dobzhansky-muller incompatibilites contributing to the sterility of hybrids between mimulus guttatus and m. nasutus. Evolution 55: 1932–1942.
Garavito A, Guyot R, Lozano J, Gavory F, Samain S, Panaud O et al. (2010). A genetic model for the female sterility barrier between asian and african cultivated rice species. Genetics 185: 1425–1440.
Harushima Y, Nakagahra M, Yano M, Sasaki T, Kurata N . (2001). A genome-wide survey of reproductive barriers in an intraspecific hybrid. Genetics 159: 883–892.
Harushima Y, Nakagahra M, Yano M, Sasaki T, Kurata N . (2002). Diverse variation of reproductive barriers in three intraspecific rice crosses. Genetics 160: 313–322.
Kubo T, Yamagata Y, Eguchi M, Yoshimura A . (2008). A novel epistatic interaction at two loci causing hybrid male sterility in an inter-subspecific cross of rice (Oryza sativa L.). Genes Genet Syst 83: 443–453.
Kubo T, Yoshimura A, Kurata N . (2011). Hybrid male sterility in rice is due to epistatic interactions with a pollen killer locus. Genetics 189: 1083–1092.
López-Fernández H, Bolnick DI . (2007). What causes partial f1 hybrid viability? incomplete penetrance versus genetic variation. PLoS One 2: e1294.
Lu H, Romero-Severson J, Bernardo R . (2002). Chromosomal regions associated with segregation distortion in maize. Theor Appl Genet 105: 622–628.
Luo L, Xu S . (2003). Mapping viability loci using molecular markers. Heredity 90: 459–467.
Luo L, Zhang YM, Xu S . (2004). A quantitative genetics model for viability selection. Heredity 94: 347–355.
Moyle LC, Graham EB . (2006). Genome-wide associations between hybrid sterility qtl and marker transmission ratio distortion. Mol Biol Evol 23: 973–980.
Moyle LC, Nakazato T . (2008). Comparative genetics of hybrid incompatibility: sterility in two solanum species crosses. Genetics 179: 1437–1453.
Moyle LC, Nakazato T . (2009). Complex epistasis for dobzhansky–muller hybrid incompatibility in solanum. Genetics 181: 347–351.
Muller H . (1942). Isolating mechanisms, evolution, and temperature. Biol. Symp 6: 71–125.
Orr HA, Irving S . (2001). Complex epistasis and the genetic basis of hybrid sterility in the Drosophila pseudoobscura Bogota-USA hybridization. Genetics 158: 1089–1100.
Ouyang Y, Liu YG, Zhang Q . (2010). Hybrid sterility in plant: stories from rice. Curr Opin Plant Biol 13: 186–192.
Phillips PC . (1998). The language of gene interaction. Genetics 149: 1167–1171.
Sharma HC, Gill BS . (1983). Current status of wide hybridization in wheat. Euphytica 32: 17–31.
Song X, Qiu SQ, Xu CG, Li XH, Zhang Q . (2005). Genetic dissection of embryo sac fertility, pollen fertility, and their contributions to spikelet fertility of intersubspecific hybrids in rice. Theor Appl Genet 110: 205–211.
Vogl C, Xu S . (2000). Multipoint mapping of viability and segregation distorting loci using molecular markers. Genetics 155: 1439–1447.
Wang C, Zhu C, Zhai H, Wan J . (2005). Mapping segregation distortion loci and quantitative trait loci for spikelet sterility in rice (Oryza sativa L.). Genet Res 86: 97–106.
Zhu C, Zhang YM . (2007). An EM algorithm for mapping segregation distortion loci. BMC Genet 8: 82.
Acknowledgements
This work was supported by the Natural Science Foundation of Hubei Province, People’s Republic of China (Grant No. 2008CDB082). We thank Susan R McCouch for sharing the RFLP mapping data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Xie, S., Chen, J. & Walsh, B. Genetic mapping of sterile genes with epistasis in backcross designs. Heredity 112, 165–171 (2014). https://doi.org/10.1038/hdy.2013.88
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2013.88
