
Abstract
There has been much debate concerning whether cis-regulatory or coding changes are more likely to produce evolutionary innovation or adaptation in gene function, but an additional complication is that some genes can dramatically diverge through alternative splicing, increasing the diversity of gene function within a locus. The fruitless gene is a major transcription factor with a wide range of pleiotropic functions, including a fundamental conserved role in sexual differentiation, species-specific morphology and an important influence on male sexual behaviour. Here, we examine the structure of fruitless in multiple species of Drosophila, and determine the patterns of selective constraint acting across the coding region. We found that the pattern of selection, estimated from the ratio of non-synonymous to synonymous substitutions, varied considerably across the gene, with most regions of the gene evolutionarily conserved but with several regions showing evidence of divergence as a result of positive selection. The regions that showed evidence of positive selection were found to be localised to relatively consistent regions across multiple speciation events, and are associated with alternative splicing. Alternative splicing may thus provide a route to gene diversification in key regulatory loci.
Similar content being viewed by others


Introduction
The nature of the genes that cause important evolutionary change is much debated (Stern, 2000; Carroll, 2005; Hoekstra and Coyne, 2007; Stern and Orgogozo, 2008, 2009). Recently, this has often focused on whether cis-regulatory or coding changes are more likely to produce evolutionary innovation or adaptation. Currently the data to test this are not conclusive either way (Hoekstra and Coyne, 2007; Stern and Orgogozo, 2008); however, it does appear that cis-regulatory changes may be more likely to underlie differences above the species level (Stern and Orgogozo, 2008). Despite the debate, it is clear that both coding and non-coding changes can cause species differences. For example, the evolution of key odorant receptor loci may underlie ecological speciation in Drosophila sechellia (Matsuo et al., 2007), whereas changes in the expression of genes involved in sexually dimorphic pheromonal production may influence sexual isolation in the same species group (Shirangi et al., 2009).
The argument in favour of cis-regulatory changes is based primarily on the idea that changes in cis-regulatory regions are less likely to suffer from the negative effects of pleiotropy, due to their modular nature (Carroll, 2005; Stern and Orgogozo, 2008). However, there are alternative genetic mechanisms that may ameliorate the constraint imposed by the pleiotropy associated with coding changes, for example, neofunctionalism resulting from gene duplication (Lynch et al., 2001; Innan and Kondrashov, 2010). Another, much less well-studied mechanism is alternative splicing (Long et al., 2003). Gene duplication and alternative splicing allow gene diversification by reducing the functional constraint on a gene (Graveley, 2001; Chothia et al., 2003). Alternative splicing and gene duplication appear to be negatively correlated at a genomic level (Kopelman et al., 2005; Talavera et al., 2007; Jin et al., 2008), suggesting that gene duplication and alternative splicing may be alternative evolutionary mechanisms influencing gene diversity (Kopelman et al., 2005). Although both processes reduce the amount of functional constraint on a sequence, allowing changes in gene product and expression, the location and type of the changes involved have been found to be different. Substitutions occurring within alternatively spliced genes are both more localised (mainly in those exons being alternatively spliced) and less conservative than those in genes that have been duplicated (Talavera et al., 2007). The gene fruitless (fru) is an alternatively spliced transcription factor that has been identified in a broad range of insect groups (Salvemini et al., 2010), including Orthoptera (Ustinova and Mayer, 2006; Boerjan et al., 2011), Blattodea (Clynen et al., 2011), Hymenoptera (Bertossa et al., 2009) and Diptera (Ryner et al., 1996; Gailey et al., 2006; Salvemini et al., 2009; Sobrinho and de Brito, 2010; Salvemini et al., 2013). fru is a pleiotropic gene with at least two major functions: one that controls male sexual behaviour and another that is essential for viability in both sexes. All Fru proteins are putative transcription factors containing a common BTB (protein:protein interaction) N-terminal domain, a connector region and, through alternative splicing, one of four C-terminal Zn finger DNA-binding domains (A, B, C and D). Transcripts from the most distal fru promoter, P1, undergo sex-specific alternative splicing and encode the male-specific FruM proteins that only differ from the common isoforms by the addition of 101 amino acids at the N-terminus. These male-specific putative transcription factors determine many of the neuronal substrates for sexual behaviour in the male central nervous system (Figure 1) (Ito et al., 1996; Ryner et al., 1996).

The structure and splicing pattern of the fruitless gene in D. melanogaster. P1 promoter mRNA transcripts are sex-specifically spliced at the 5′-end, resulting in the inclusion of the S exon and the addition of 101 amino acids (yellow) to male-specific isoforms (FruM) and the inclusion of a premature stop codon in females (UAA). Alternative splicing at the 3′-end of transcripts produced from the sex-specific P1 promoter and non-sex-specific P2-4 promoters results in the inclusion of alternative DNA-binding domains A (purple), B (orange), C (green) or D (brown). All isoforms contain the BTB domain (blue) and connector region (grey). Common exons C1-5 are included in fruA/B/C isoforms, whereas the fruD isoform includes exons C1–C4. Untranslated regions (UTRs) are shown in white and translation start codons are indicted (ATG).
The high level of pleiotropy associated with fru suggests that it should be evolutionarily conserved (Wilkins, 1995; Billeter et al., 2006a). Such conservation was shown by the ability of the Anopheles gambiae ortholog of fru to function when ectopically expressed in D. melanogaster resulting in the production of the fru-dependent male-specific muscle of Lawrence (Gailey et al., 2006). As A. gambiae and D. melanogaster have been separated for ∼250 mya (Gaunt and Miles, 2002; Zdobnov et al., 2002, Gailey et al. (2006) concluded that fru has been functionally conserved across this time period. This has been further emphasised with the finding that RNAi-mediated knockdown of fru extinguishes male courtship in the cockroach Blattella germanica, suggesting that the large role fru has in the production of male sexual behaviours has been conserved for at least a large portion of insect evolution (Clynen et al., 2011). Despite this, many of the courtship behaviours influenced by fru are known to be species-specific, and fru has been implicated as a potential candidate gene for species-specific divergence in QTL (quantitative trait loci) studies (Gleason and Ritchie, 2004; Lagisz et al., 2012). Furthermore, a recent study of the fru connector region using three species of fruit fly (Genus: Anastrepha) found evidence of positive selection based on both sequence differences and population gene frequencies, suggesting that fru may contribute to species-specific differences in male courtship behaviour of Anastrepha species (Sobrinho and de Brito, 2010).
This highlights an intriguing conundrum about the widespread use of candidate genes in evolutionary biology; important genes would be expected to be under selective constraint, yet to be important to adaptation, such genes must evolve rapidly between species. The candidate gene approach has proven very successful in numerous studies of species differences (Martin and Orgogozo, 2013), including studies of behaviour (Fitzpatrick et al., 2005). fru provides an example of such a gene: on the one hand, fru is known to be a highly pleiotropic essential gene for both sexes, suggesting it should be highly conserved. On the other hand, fru has been implicated in the production of behaviour, which is typically species-specific. One possible resolution to this is that the alternative splicing of the exons in fru may allow some exons to accumulate changes that alter species-specific behaviour, while other exons are conserved to maintain their essential functions. To address this we have conducted an analysis of the fru-coding region from 18 species of sequenced Drosophila. We examine (i) the pattern of sequence variability across exons of fru between Drosophila species, (ii) what proportion, if any, of such variability is due to positive selection and (iii) if divergently selected regions of fru specifically occur in the alternatively spliced exons.
Materials and methods
Drosophila species
The Drosophila genome assemblies used in this paper were downloaded from the following websites in July 2012:
-
1
D. melanogaster (v. 5.47) from FlyBase (http://flybase.org/).
-
2
D. simulans, D. sechellia D. yakuba, D. erecta, D. ananassae, D. persimilis, D. pseudoobscura, D. willistoni, D. virilis, D. mojavensis and D. grimshawi from http://rana.lbl.gov/drosophila/assemblies.html (CAF1, comparative analysis freeze (1). Further information on these genome assemblies is available from Drosophila 12 Genomes Consortium (2007). In addition, the B exon for D. simulans was not available from the CAF1 assembly due to sequence failure in this region, and so the sequence for this exon was obtained from Genbank (accession number: GI: 111258132). We also re-sequenced the C exon for D. simulans and D. sechellia (see below) as these regions were also unavailable from the CAF1 assembly.
-
3
D. bipectinata, D. kikkawai, D. elegans, D. eugracilis, D. ficusphila, D. rhopaloa, D. biarmipes and D. takahashii from https://www.hgsc.bcm.edu/content/drosophila-modencode-project. The sequencing was provided by Baylor College of Medicine Human Genome Sequencing Centre.
Re-sequencing assembly gaps in the fru locus
To obtain the sequence of the C exon of fru for D. simulans and D. sechellia genomic DNA was extracted from inbred lines of D. simulans (f2;nt, pm; st, e, kindly provided by Jerry Coyne) and D. sechellia (David4A, kindly provided by Jean R. David) (see Gleason and Ritchie, 2004) using the single fly prep method developed by Gloor et al. (1993). The resulting extractions were then amplified via PCR using the following primers designed from the orthologous region in D. melanogaster: 5′-GACGGGCTGTTGTGTGTTC-3′ and 5′-CACGCCCTTAAATGGATGA-3′. The PCR products from these reactions were then Sanger sequenced using Dundee Sequencing Services (www.dnaseq.co.uk), the consensus sequences of which were then submitted to Genbank (accession numbers: KF005597 and KF005598 for D. simulans and D. sechellia, respectively).
Annotation of the fru orthologs in Drosophila species
Annotation of the orthologs of D. melanogaster fruitless (fru, CG14307) gene was performed for the other Drosophila species using a combination of BLAST (Altschul et al., 1990), GeneWise (Birney et al., 2004) and manual curation. Available amino-acid sequences of the proteins encoded by the fruitless (FlyBase, FBpp0083060–67 and FBpp00839355-59) of D. melanogaster were used as the queries in TBLASTN search of each of the other Drosophila species’ genomic DNA in turn. The worst scoring alignments were discounted. For the remainder, the genomic DNA involved in the alignment, with flanking regions, was extracted using a simple BioPerl script (Stajich et al., 2002). Provisional gene structures were predicted automatically by realigning the D. melanogaster proteins and the genomic region using GeneWise. Finally, coordinates of exons in the GeneWise predictions were corrected manually. This was necessary to obtain a realistic gene structure where the protein sequence diverged from that of the D. melanogaster protein in the region of a start, stop or splice site, causing the GeneWise model to truncate the exon. Thus, the loci structure and protein-coding exons were identified across 18 species of Drosophila. The D. persimilis and D. rhopaloa genome assemblies were found to have poor coverage of the region that includes fruitless, so we excluded these species from our analysis. The size of fru orthologs was defined as the sequence from the transcription start site in promoter 1 (P1) to the end of the C exon (Figure 1, Supplementary Table 1).
Sequence analysis
The protein-coding sequences of fru were multiply aligned using ClustalW (Thompson et al., 1994) on translations, followed by Protal2dna (K. Schuerer, C. Letondal; http://bioweb.pasteur.fr) to obtain a codon alignment for use in PAML (below). Pairwise nucleotide identity values for the codon aligned sequences were obtained using the Geneious program (version 5.6.6. available from www.geneious.com).
The M0 model of codeml in the PAML computer package (Yang, 1997) was used to determine overall selective constraint acting on the fru protein-coding exons through estimation of the ratio of the normalised non-synonymous substitution rate (dN) to normalised synonymous substitution rate (dS) or ω=dN/dS. ω>1 is considered to be strong evidence of positive selection for amino-acid replacements, whereas ω≈0 indicates purifying selection (Yang and Bielawski, 2000).
The alternative splicing of fruitless produces a number of well-defined transcripts in D. melanogaster of which the following were tested for evidence of positive selection across all of the species: the set of transcripts that consist of C1–C5 exons and one of the 3′-alternatively spliced exon ends (either A (Fru-RI, FBtr0083648), B (Fru-RK, FBtr0083650) or C (Fru-RF, FBtr0083644)), the transcript that includes exons C1–C4 and exon D (Fru-RD, FBtr0083647), the C1–C5 exons alone (Fru-RA, FBtr0083646), and the three male-specific fru transcripts, which include the C1–C5 exons, sex-specific N-terminus (S) and one of the 3′ alternatively spliced exon ends (either A (FruMA), B (FruMB) or C (FruMC)) (Figure 1). In addition, we also tested exon S separately.
To test for evidence of positive selection on the fru products, we used M7 vs M8 and M8a vs M8 site-based model comparisons in PAML (Yang, 1997). Models M7 and M8a are null models, which do not allow any sites to have ω>1. M8 has the additional parameter of a class of sites (p1) which allow ω>1. Models are compared by a log-likelihood ratio test, LRT (LRT=−2 times the difference in log-likelihood tested against a χ2-distribution with the number of degrees of freedom equal to the number of additional random effects). It should be noted that the use of two degrees of freedom for the M8 vs M7 comparisons and one degree of freedom for the M8a vs M8 comparisons is considered conservative (Swanson et al., 2003; Wong et al., 2004).
Site-based models average the value of ω over all of the branches in the tree meaning such tests lack power if selection has been concentrated on only a few branches. One could apply branch-based or branch-site-based models of selection, which allow the value of ω to vary between linages. A problem with this method is that any such divisions must be applied a priori and it is unclear why we would expect selection on fru to differ among Drosophila linages. As a result, we did not apply any branch or branch site models to our data. The tree provided to PAML for selection analyses was produced using trees from Da Lage et al., 2007 and Drosophila 12 Genomes Consortium 2007 (Supplementary Figure 1).
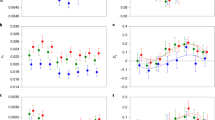
In order to obtain a visual indication of the regions of fru, showing the highest values of ω, pairwise comparison of the values of ω along the fru-coding regions was conducted between D. melanogaster and the other sequenced melanogaster group species (D. elegans, D. eugracilis, D. ficusphila, D. biarmipes, D. takahashii, D. yakuba, D. erecta, D. sechellia and D. simulans) using a sliding window. The size of the window for calculating ω for comparisons using D. elegans, D. eugracilis, D. ficusphila, D. biarmipes, D. takahashii, D. yakuba and D. erecta was 102 bp (that is, the fru alignment was split into 102 bp ‘windows’, from which a value of ω was calculated). Windows that did not show any synonymous changes were combined with the following window to allow calculation of ω. For comparisons using the more closely related D. sechellia and D. simulans a 408-bp window was used, because there were a large number of regions with no synonymous changes. This 408 bp window was then moved by 102 bp to allow the regions of fru with the highest values of ω to be visualised. To avoid analysing any chimeric sequences, values of ω for each of the alternatively spliced exons (S, A, B, C and D) were calculated separately before concatenation to produce Figures 2 and 3.

Values of dN/dS (ω) between D. melanogaster and D. simulans, D. sechellia, D. erecta and D. yakuba across the coding region of fruitless. Values for each point represent the average dN/dS value for either a 102 bp window for D. erecta and D. yakuba or a 408-bp window for D. sechellia and D. simulans.

Values of dN/dS (ω) between D. melanogaster and D. takahashi, D. biarmipes, D. eugracilis, D. fisusphila and D. elegans across the coding region of fruitless. Values for each point represent the average dN/dS value in a 102-bp window.
Results
Genomic location of the fru locus
The gene fruitless is located on the right arm of the third chromosome (3R) in the D. melanogaster genome, spanning nearly 130 kbp, in cytological position 91A7-91B3 with genes CG31122 and CG7691 located up and downstream of fru, respectively. We identified single copy orthologs of fru in 17 other Drosophila species. Only D. simulans, D. yakuba and D. pseudoobscura genomes are localised to chromosomes, the remainder are only available as scaffolds. We identified the location of the fru locus in each species and an approximate length of the region encompassing the fru exons (Supplementary Table 1). In D. simulans and D. yakuba, fru is located on the right arm of the third chromosome (as in D. melanogaster), and on the second Muller element in D. pseudoobscura (homologous to the 3R of D. melanogaster) (Powell, 1997). The total length of the fru locus varies between species from 117 kbp in D. bipectinata and 167 kbp in D. mojavensis (Supplementary Table 1). Local synteny of genes appears to be conserved as all but one of the fru orthologs identified in this study are flanked by the orthologs of CG31122 and CG7691. The fru ortholog of D. kikkawai is flanked by CG31122 but not CG7691. This, however, is unlikely to represent a change in local synteny, but rather is a result of fru occurring near the end of the assembled scaffold.
Organisation and structure of fru
Common exons
Across Drosophila species, we identified exons C1–C5 and reconstructed the exon–intron structure of this region. Putative splice donor and acceptor sites are in agreement with the consensus motifs (Mount et al., 1992). The exons C1, C2 and part of C3 encode for BTB/POZ domains and the remainder of C3, C4 and C5 encode for the ‘connector’ that joins BTB and 3′ zinc-finger domains. The Fru BTB domain is a highly conserved ∼120 amino-acid long domain, found in many other D. melanogaster transcription factors (Zollman et al., 1994; Bonchuk et al., 2011). Across the species we found that the C1 and C2 exons are highly conserved, with pairwise nucleotide identity of 94% and few amino-acid substitutions across all species (two sites in C1 and 1 site in C2). The nucleotide and amino-acid similarity is reduced in the C3, C4 and C5 exons with pairwise nucleotide identity values of 79%, 84% and 83%, respectively.
Alternative 3′-ends-zinc-finger domains
A schematic of alternative splicing of the fru exons is presented in Figure 1. There are four main alternative 3′-exons: A, B, C and D. Exons A, B and C each contain two C2H2 zinc-finger-binding domains (Ito et al., 1996; Ryner et al., 1996; Usui-Aoki et al., 2000). Manual inspection of the exon D alignment identified a pair of conserved cysteine and histidine residues separated by a motif of 28 amino acids (consensus sequence: CRHC RKWSGELADIRTSFVEGNSNFRLEIVNH HNKCKSH—cysteine and histidine motifs underlined). This is a significant departure from the consensus ‘finger’ sequences (Wolfe et al., 2000) suggesting that exon D encodes for either an atypical zinc-finger domain, a non-functional domain or a domain with novel structure. The zinc-finger motifs of exons A, B and C have no amino-acid substitutions across all species and the proposed zinc-finger motif of the D exon has only two amino-acid sites, which vary between these species. Pairwise nucleotide identity values vary for the four alternative 3′-exons, with exons A and D showing less sequence conservation across species than exons B and C (pairwise nucleotide identity values for exons A, B, C and D: 62%, 82%, 76% and 71%, respectively).
Alternative 5′-sex-specific exon
The alternatively spliced exon S was found to be similar across species with a pairwise nucleotide identity value of 77%. In addition, the three transformer (tra/tra2) binding sites in the S exon UTR were also found to be highly conserved (pairwise nucleotide identity value of sites 96.4%, 97.2% and 88.6%, respectively) (see Supplementary Figures 2 and 3 for alignments).
Selection analysis
Across the whole-coding region of fru the value of ω was 0.107, implying purifying selection is acting; however, the value of ω varies widely across the gene. Selective constraints on the region coding for BTB domain are very strong (ωBTB=0.013), while the strength of purifying selection acting on the C3–C5 exons encoding the ‘domains connector’ is weaker, with an average ω=0.064. Purifying selection on 80 amino acids that include the zinc-finger motifs on exons A, B, C and D is very strong (ωZnF-A=0.00184; ωZnF-B=0.00010; ωZnF-C=0.00375; ωZnFD=0.01805) with weaker constraint acting on the rest of the exon (ωA=0.219; ωB=0.077; ωC=0.186; ωD=0.145). Selective constraint across the region coding for the 5′ sex-specifically spliced exon S was also found to be mainly purifying (ωS=0.074).
Comparison of the nested models M7 and M8 across the whole-coding region of fru found M8 to be a significantly better fit (P=0.00001) with 3.4% of sites (p1=0.03414, ω=1.46311) under positive selection. The more stringent test for positive selection (the comparison of the M8a and M8 models) also found M8 to be a better fit (P=0.005). Comparison of M7 and M8 found M8 to be a significantly better fit for most of the known transcripts (Table 1); however, M8 was a better fit for only three of transcripts when compared with M8a. These contained either exon A (Fru-RI and FruMA) or exon D (Fru-RD) indicating positive selection on these regions (Table 1). For those transcripts, the proportion of sites under positive selection (p1) was around 4% (Fru-RI: p1=0.0383, ω=1.412; FruMA: p1=0.0382, ω=1.454; Fru-RD: p1=0.0357, ω=1.683) (Table 1). Transcripts containing other exons either showed the M8 model to be a better fit than M7 but not M8a (exons B and C) or M8 was not a better fit than M7 (C1–C5, containing only the BTB domain and the connector), implying these regions are evolving neutrally or under purifying selection, respectively. The M8 model was also found to not be a better fit than M7 for exon S (P=0.656, Table 1) implying this exon is also evolving under purifying selection.
Pairwise sliding window comparisons of fru across the melanogaster group species (Figures 2 and 3) shows values of ω are elevated in similar areas in each of the pairwise comparisons: around the 5′-end of the A exon, in line with the finding that transcripts containing the A exon are under positive selection (Table 1). There is evidence for saturation, because values of ω for species more distant to D. melanogaster have lower peaks of ω, probably as a result of a large number of synonymous changes rather than a lack of non-synonymous changes (Figure 4). The pairwise sliding window comparisons, however, did not show peaks in the region containing exon D, despite evidence for positive selection on transcripts containing this exon. An explanation for this may be that the positively selected changes in exon D are less localised than in exon A, and that, unlike exon A, the putative zinc-finger for exon D is in the middle of this exon, which may make sites of diversifying selection more difficult to visualise.

Values of dN and dS for melanogaster group species from pairwise comparisons with D. melanogaster.
Discussion
Divergence during speciation is thought to be driven by strong selection (Coyne and Orr, 2004; Rundle and Nosil, 2005), thus such divergence would be expected to leave a signature of an excess of non-synonymous substitutions (dN) between closely related species. However, the increasing availability of genome projects and focussed studies of gene families are finding that relatively few genes show elevated dN in genomic comparisons (Drosophila 12 Genomes Consortium, 2007; Ellegren et al., 2012). Relaxed selection, especially following gene duplication, is undoubtedly also important to the evolution of new gene functions and species differences. fru is a gene with highly pleiotropic functions, some of which are essential for viability in both sexes (Anand et al., 2001; Song et al., 2002; Song and Taylor, 2003). Previous studies have suggested that fru should be evolutionarily conserved (Wilkins, 1995; Gailey et al., 2006; Salvemini et al., 2009; Clynen et al., 2011), yet it has also been implicated in the production of sexually dimorphic behaviour, which is known to change rapidly between species (Mendelson and Shaw, 2005; Kraaijeveld et al., 2011). In addition, fru has also been implicated as a potential candidate gene for the production of species-specific behaviour differences (Gleason and Ritchie, 2004; Sobrinho and de Brito, 2010). The alternative splicing of fru may offer a resolution of this apparent contradiction, if some exons accumulate changes that alter species-specific behaviour, while other exons remain conserved to maintain their essential functions. This predicts that different transcripts of the same gene should have rather different evolutionary rates and show variation in the relative rate of non-synonymous substitutions.
Positive selection is restricted to alternatively spliced exons
We found evidence of positive selection acting on a small but significant number of sites in the fru-coding region (Table 1). These sites are restricted to transcripts containing alternatively spliced exons A or D. In contrast, alternatively spliced exons B and C did not show evidence of positive selection, and appear to be governed primarily by purifying selection with a small proportion of neutrally evolving sites (Table 1). The male-specific alternatively spliced exon S and common coding regions of fru transcripts also showed no evidence of positive selection and appear to be under strong selective constraints.
These findings raise clear predictions concerning the functional importance of different transcripts, which, for example, could be tested by mutagenesis or selective introgression experiments. As transcripts containing exons B and C were found to be conserved, we hypothesise that they are responsible for the essential functions of fru, whereas transcripts containing exons A and D are more likely be involved in non-essential functions, which may contribute to phenotypic differences between species. As exon D does not appear to be included in fru isoforms controlling male sexual behaviour (Billeter et al., 2006b), we further hypothesise that sequence variation in isoforms containing exon A, could influence species-specific differences in male sexual behaviour. We know from molecular genetic studies, that fru exploits these multiple isoforms through spatial and temporal expression of either a single, or a combination of isoforms enabling specific phenotypic outcomes. For instance, the production of serotonergic neurons in the central nervous system that innervate the male reproductive system depends on the expression of FruMB and FruMC isoforms and not the FruMA isoform (Billeter et al., 2006b).
Our finding of positive selection in alternatively spliced exons at the 3′-end of fru raises the question of why no positive selection was found in alternatively spliced exon S towards the 5′-end of fru. A potential solution is that, although exon S is alternatively spliced, it is either present or absent in fru transcripts (that is, there is no alternative exon to S, isoforms vary only in the presence or absence of exon S). This means that, unlike at the 3′-end of fru, the alternative splicing of exon S does not provide redundancy at the 5′-end of fru, and thus does not provide any reduction in selective constraint for this exon.
Our finding, that positively selected changes are localised to alternatively spliced exons, is in broad agreement with previous studies that have shown that typically there are a greater number of positively selected changes in alternatively spliced exons than in constitutively spliced exons (Ermakova et al., 2006; Ramensky et al., 2008; Hughes, 2011). This suggests that alternative splicing may provide a general mechanism for the evoultion of noveliy in otherwise conserved genes. In contrast, a previous study looking at the patterns of selection on fru in Anastrepha fruit flies (Sobrinho and de Brito, 2010) found evidence for positive selection on constitutively spliced exon C3. We did not find evidence of positive selection in this region, however, it is not known if positive selection also occurs in the alternatively spliced regions of Anastrepha fru as these regions are not currently available for study, making direct comparisons with our study difficult.
Positive selection on alternatively spliced exons presumably arises due to changes in protein structure. However, splicing regulation occurs via changes in exonic-splicing regulators (ESRs), which are presumably themselves under selection. ESRs are typically short sequences (usually hexamers) within coding regions, which enhance or suppress splicing. As ESR motifs are regulatory in function, functional changes will not necessarily be detected by dN/dS style analyses. Selected changes in ESRs should not favour non-synonymous changes over synonymous changes (synonymous changes, in fact, should be more likely to avoid potentially deleterious changes in protein sequence). This combined with the fact that ESRs are typically quite short, means that selection for changes in splicing regulation via ESRs is unlikely to be found by this analysis, so the evidence for positive selection found in this study is more likely to reflect selection for changes in the protein sequence.
How could the positive selection detected in some transcripts of fru act to alter traits, including distinct behaviours? As fru is a transcription factor, sequence changes could either cause change in the target loci it binds to, or it could alter the expression of a similar suite of downstream loci. Our data perhaps suggest that the latter is more likely; the zinc-finger motifs of all the 3′-alternatively spliced exons (A, B, C and D) are highly conserved. This suggests that the positive selection detected is unlikely to be changing the sites the transcription factor binds to between species. As transcription factors typically interact with several proteins while binding DNA, changes to the amino-acid sequence outside the zinc-finger may affect the efficiency with which the transcription factor is able to bind to the target DNA and/or influence the way the transcription factor interacts with other proteins (Locker, 2001). As such, the changes in exon A and D may influence the regulation of downstream genes to which the zinc-finger binds. Currently, the genes directly regulated by fru are unknown (Villella and Hall, 2008), however, as fru is known to be a major gene in the sex determination cascade, the changes in fru found by this study may influence the expression of a large number of downstream targets (Baker et al., 2007).
Owing to fru’s position in the sex determination pathway and the role it has in the shaping of male sexual behaviour, these results suggest that fru may be acting as a ‘hotspot gene’ for the evolution of male sexual traits. Hotspot genes are those genes which are able to incur a disproportionate number of evolutionary important mutations for a trait: mutations, which cause a large enough phenotypic change for selection to act upon and that are able to be positive selected due to limited negative pleiotropy (Stern, 2000; Stern and Orgogozo, 2009; Martin and Orgogozo, 2013). Stern and Orgogozo (2009) suggest that such hotspot genes will contribute disproportionally to the evolution of differences between species. Of course, numerous high resolution QTL studies of species differences will be required to assess the likelihood of a disproportionate role of individual loci in species differences. Stern and Orgogozo (2009) also suggest that regions of a gene, which experience less pleiotropy would be more likely to accumulate evolutionary relevant mutations. They suggested this in the context of cis-regulatory vs coding mutations whereby cis-regulatory mutations would be more likely to accumulate changes (Stern, 2000; Carroll, 2005; Hoekstra and Coyne, 2007; Stern and Orgogozo, 2008; Stern and Orgogozo, 2009). The same might be true for alternatively spliced regions, which are likely to experience less pleiotropy than common coding regions due to the functional redundancy the production of alternative transcripts provides. Our findings are consistent with this: we found that positively selected changes in fru had accumulated in two of the alternatively spliced exons, showing that alternative splicing may impact a gene’s ability to accumulate evolutionary relevant mutations. In many ways, this is similar to the role of neofunctionalisation of recent duplicate loci in the generation of evolutionary novelty (Lynch and Conery, 2000). The widespread incidence of alternative splicing in plasticity, gene function and adaptation is starting to be understood, but how this will contribute to adaptive divergence and ultimately speciation is only beginning to be explored (Ast, 2004; Harr and Turner, 2010).
Data Archiving
This paper contains two new sequences which are available from Genbank, the accession numbers of which are given below.

References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ . (1990). Basic local alignment search tool. J Mol Biol 215: 403–410.
Anand A, Villella A, Ryner LC, Carlo T, Goodwin SF, Song HJ et al. (2001). Molecular genetic dissection of the sex-specific and vital functions of the Drosophila melanogaster sex determination gene fruitless.. Genetics 158: 1569–1595.
Ast G . (2004). How did alternative splicing evolve? Nat Rev Genet 5: 773–782.
Baker DA, Meadows LA, Wang J, Dow JA, Russell S . (2007). Variable sexually dimorphic gene expression in laboratory strains of Drosophila melanogaster. BMC Genomics 8: 454.
Bertossa RC, van de Zande L, Beukeboom LW . (2009). The fruitless gene in Nasonia displays complex sex-specific splicing and contains new zinc finger domains. Mol Biol Evol 26: 1557–1569.
Billeter JC, Rideout EJ, Dornan AJ, Goodwin SF . (2006a). Control of male sexual behavior in Drosophila by the sex determination pathway. Curr Biol 16: R766–R776.
Billeter JC, Villella A, Allendorfer JB, Dornan AJ, Richardson M, Gailey DA et al. (2006b). Isoform-specific control of male neuronal differentiation and behavior in Drosophila by the fruitless gene. Curr Biol 16: 1063–1076.
Birney E, Clamp M, Durbin R . (2004). GeneWise and genomewise. Genome Res 14: 988–995.
Boerjan B, Tobback J, De Loof A, Schoofs L, Huybrechts R . (2011). fruitless RNAi knockdown in males interferes with copulation success in Schistocerca gregaria. Insect Biochem Mol Biol 41: 340–347.
Bonchuk A, Denisov S, Georgiev P, Maksimenko O . (2011). Drosophila BTB/POZ domains of ‘ttk group’ can form multimers and selectively interact with each other. J Mol Biol 412: 423–436.
Carroll SB . (2005). Evolution at two levels: on genes and form. PLoS Biol 3: 1159–1166.
Chothia C, Gough J, Vogel C, Teichmann SA . (2003). Evolution of the protein repertoire. Science 300: 1701–1703.
Clynen E, Ciudad L, Belles X, Piulachs MD . (2011). Conservation of fruitless, role as master regulator of male courtship behaviour from cockroaches to flies. Dev Genes Evol 221: 43–48.
Coyne JA, Orr HA . (2004) Speciation. Sinauer: Sunderland, MA, USA.
Da Lage JL, Kergoat GJ, Maczkowiak F, Silvain JF, Cariou ML, Lachaise D . (2007). A phylogeny of Drosophilidae using the Amyrel gene: questioning the Drosophila melanogaster species group boundaries. J Zool System Evol Res 45: 47–63.
Drosophila 12 Genomes Consortium. (2007). Evolution of genes and genomes on the Drosophila phylogeny. Nature 450: 203–218.
Ellegren H, Smeds L, Burri R, Olason PI, Backstrom N, Kawakami T et al. (2012). The genomic landscape of species divergence in Ficedula flycatchers. Nature 491: 756–760.
Ermakova EO, Nurtdinov RN, Gelfand MS . (2006). Fast rate of evolution in alternatively spliced coding regions of mammalian genes. BMC Genomics. 7: 84.
Fitzpatrick MJ, Ben-Shahar Y, Smid HM, Vet LEM, Robinson GE, Sokolowski MB . (2005). Candidate genes for behavioural ecology. Trends Ecol Evol 20: 96–104.
Gailey DA, Billeter JC, Liu JH, Bauzon F, Allendorfer JB, Goodwin SF . (2006). Functional conservation of the fruitless male sex-determination gene across 250Myr of insect evolution. Mol Biol Evol 23: 633–643.
Gaunt MW, Miles MA . (2002). An insect molecular clock dates the origin of the insects and accords with palaeontological and biogeographic landmarks. Mol Biol Evol 19: 748–761.
Gleason JM, Ritchie MG . (2004). Do quantitative trait loci (QTL) for a courtship song difference between Drosophila simulans and D. sechellia coincide with candidate genes and intraspecific QTL? Genetics 166: 1303–1311.
Gloor GB, Preston CR, Johnsonschlitz DM, Nassif NA, Phillis RW, Benz WK et al. (1993). Type-I repressors of P-element mobility. Genetics 135: 81–95.
Graveley BR . (2001). Alternative splicing: increasing diversity in the proteomic world. Trends Genet 17: 100–107.
Harr B, Turner LM . (2010). Genome-wide analysis of alternative splicing evolution among Mus subspecies. Mol Ecol 19: 228–239.
Hoekstra HE, Coyne JA . (2007). The locus of evolution: Evo devo and the genetics of adaptation. Evolution 61: 995–1016.
Hughes AL . (2011). Runaway evolution of the male-specific exon of the doublesex gene in Diptera. Gene 472: 1–6.
Innan H, Kondrashov F . (2010). The evolution of gene duplications: classifying and distinguishing between models. Nat Rev Genet 11: 97–108.
Ito H, Fujitani K, Usui K, ShimizuNishikawa K, Tanaka S, Yamamoto D . (1996). Sexual orientation in Drosophila is altered by the satori mutation in the sex-determination gene fruitless that encodes a zinc finger protein with a BTB domain. Proc Natl Acad Sci USA 93: 9687–9692.
Jin L, Kryukov K, Clemente JC, Komiyama T, Suzuki Y, Imanishi T et al. (2008). The evolutionary relationship between gene duplication and alternative splicing. Gene 427: 19–31.
Kopelman NM, Lancet D, Yanai I . (2005). Alternative splicing and gene duplication are inversely correlated evolutionary mechanisms. Nature Genet. 37: 588–589.
Kraaijeveld K, Kraaijeveld-Smit FJL, Maan ME . (2011). Sexual selection and speciation: the comparative evidence revisited. Biol Rev 86: 367–377.
Lagisz M, Wen SY, Routtu J, Klappert K, Mazzi D, Morales-Hojas R et al. (2012). Two distinct genomic regions, harbouring the period and fruitless genes, affect male courtship song in Drosophila montana. Heredity 108: 602–608.
Locker J . (2001) Transcription Factors. Academic Press: San Diego, CA, USA.
Long M, Betran E, Thornton K, Wang W . (2003). The origin of new genes: glimpses from the young and old. Nat Rev Genet 4: 865–875.
Lynch M, Conery JS . (2000). The evolutionary fate and consequences of duplicate genes. Science 290: 1151–1155.
Lynch M, O'Hely M, Walsh B, Force A . (2001). The probability of preservation of a newly arisen gene duplicate. Genetics 159: 1789–1804.
Martin A, Orgogozo V . (2013). The Loci of repeated evolution: a catalog of genetic hotspots of phenotypic variation. Evolution 67: 1235–1250.
Matsuo T, Sugaya S, Yasukawa J, Aigaki T, Fuyama Y . (2007). Odorant-binding proteins OBP57d and OBP57e affect taste perception and host-plant preference in Drosophila sechellia. PLoS Biol 5: e118.
Mendelson TC, Shaw KL . (2005). Sexual behaviour: rapid speciation in an arthropod. Nature 433: 375–376.
Mount SM, Burks C, Hertz G, Stormo GD, White O, Fields C . (1992). Splicing signals in Drosophila - intron size, information-content, and consensus sequences. Nucleic Acids Res 20: 4255–4262.
Powell JR . (1997) Progress and prospects in evolutionary biology: the Drosophila model. Oxford University Press.
Ramensky VE, Nurtdinov RN, Neverov AD, Mironov AA, Gelfand MS . (2008). Positive selection in alternatively spliced exons of human genes. Am J Human Genet 83: 94–98.
Rundle HD, Nosil P . (2005). Ecological speciation. Ecol Lett 8: 336–352.
Ryner LC, Goodwin SF, Castrillon DH, Anand A, Villella A, Baker BS et al. (1996). Control of male sexual behavior and sexual orientation in Drosophila by the fruitless gene. Cell 87: 1079–1089.
Salvemini M, D'Amato R, Petrella V, Aceto S, Nimmo D, Neira M et al. (2013). The orthologue of the fruitfly sex behaviour gene fruitless in the mosquito Aedes aegypti: evolution of genomic organisation and alternative splicing. PLoS One 8: e48554.
Salvemini M, Polito C, Saccone G . (2010). fruitless alternative splicing and sex behaviour in insects: an ancient and unforgettable love story? J Genet 89: 287–299.
Salvemini M, Robertson M, Aronson B, Atkinson P, Polito LC, Saccone G . (2009). Ceratitis capitata transformer-2 gene is required to establish and maintain the autoregulation of Cctra, the master gene for female sex determination. Int J Dev Biol 53: 109–120.
Shirangi TR, Dufour HD, Williams TM, Carroll SB . (2009). Rapid evolution of sex pheromone-producing enzyme expression in Drosophila. PLoS Biol 7: e1000168.
Sobrinho I, de Brito R . (2010). Evidence for positive selection in the gene fruitless in Anastrepha fruit flies. BMC Evol Biol 10: 293.
Song HJ, Billeter JC, Reynaud E, Carlo T, Spana EP, Perrimon N et al. (2002). The fruitless gene is required for the proper formation of axonal tracts in the embryonic central nervous system of Drosophila. Genetics 162: 1703–1724.
Song HJ, Taylor BJ . (2003). fruitless gene is required to maintain neuronal identity in evenskipped-expressing neurons in the embryonic CNS of Drosophila. J Neurobiol 55: 115–133.
Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C et al. (2002). The bioperl toolkit: Perl modules for the life sciences. Genome Res 12: 1611–1618.
Stern DL . (2000). Perspective: evolutionary developmental biology and the problem of variation. Evolution 54: 1079–1091.
Stern DL, Orgogozo V . (2008). The loci of evolution: how predictable is genetic evolution? Evolution 62: 2155–2177.
Stern DL, Orgogozo V . (2009). Is genetic evolution predictable? Science 323: 746–751.
Swanson WJ, Nielsen R, Yang QF . (2003). Pervasive adaptive evolution in mammalian fertilization proteins. Mol Biol Evol 20: 18–20.
Talavera D, Vogel C, Orozco M, Teichmann SA, de la Cruz X . (2007). The (In) dependence of alternative splicing and gene duplication. PLoS Comput Biol 3: 375–388.
Thompson JD, Higgins DG, Gibson TJ . (1994). ClustalW: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680.
Ustinova J, Mayer F . (2006). Alternative starts of transcription, several paralogues, and almost-fixed interspecific differences of the gene fruitless in a hemimetabolous insect. J Mol Evol 63: 788–800.
Usui-Aoki K, Ito H, Ui-Tei K, Takahashi K, Lukacsovich T, Awano W et al. (2000). Formation of the male-specific muscle in female Drosophila by ectopic fruitless expression. Nat Cell Biol 2: 500–506.
Villella A, Hall JC . (2008). Neurogenetics of courtship and mating in Drosophila. In: Hall JC, (ed). In Advances in Genetics Vol 62,: 67–184.
Wilkins AS . (1995). Moving up the hierarchy—A hypothesis on the evolution of a genetic sex determination pathway. Bioessays 17: 71–77.
Wolfe SA, Nekludova L, Pabo CO . (2000). DNA recognition by Cys(2)His(2) zinc finger proteins. Annu Rev Biophys Biomolec Struct 29: 183–212.
Wong WSW, Yang ZH, Goldman N, Nielsen R . (2004). Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics 168: 1041–1051.
Yang ZH . (1997). PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13: 555–556.
Yang ZH, Bielawski JP . (2000). Statistical methods for detecting molecular adaptation. Trends Ecol Evol 15: 496–503.
Zdobnov EM, von Mering C, Letunic I, Torrents D, Suyama M, Copley RR et al. (2002). Comparative genome and proteome analysis of Anopheles gambiae and Drosophila melanogaster. Science 298: 149–159.
Zollman S, Godt D, Prive GG, Couderc JL, Laski FA . (1994). The BTB domain, found primarily in zinc-finger proteins, defines an evolutionarily conserved family that includes several developmentally-regulated genes in Drosophila. Proc Natl Acad Sci USA 91: 10717–10721.
Acknowledgements
We thank Andy James and four reviewers for helpful advice on this study. This work was funded by the NERC, UK (studentship to DJP and grants to MGR and SFG).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Parker, D., Gardiner, A., Neville, M. et al. The evolution of novelty in conserved genes; evidence of positive selection in the Drosophila fruitless gene is localised to alternatively spliced exons. Heredity 112, 300–306 (2014). https://doi.org/10.1038/hdy.2013.106
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2013.106
