
Abstract
Habitat fragmentation has been shown to disrupt ecosystem processes such as plant-pollinator mutualisms. Consequently, mating patterns in remnant tree populations are expected to shift towards increased inbreeding and reduced pollen diversity, with fitness consequences for future generations. However, mating patterns and phenotypic assessments of open-pollinated progeny have rarely been combined in a single study. Here, we collected seeds from 37 Eucalyptus incrassata trees from contrasting stand densities following recent clearance in a single South Australian population (intact woodland=12.6 trees ha−1; isolated pasture=1.7 trees ha−1; population area=10 km2). 649 progeny from these trees were genotyped at eight microsatellite loci. We estimated genetic diversity, spatial genetic structure, indirect contemporary pollen flow and mating patterns for adults older than the clearance events and open-pollinated progeny sired post-clearance. A proxy of early stage progeny viability was assessed in a common garden experiment. Density had no impact on mating patterns, adult and progeny genetic diversity or progeny growth, but was associated with increased mean pollen dispersal. Weak spatial genetic structure among adults suggests high historical gene flow. We observed preliminary evidence for inbreeding depression related to stress caused by fungal infection, but which was not associated with density. Higher observed heterozygosities in adults compared with progeny may relate to weak selection on progeny and lifetime-accumulated mortality of inbred adults. E. incrassata appears to be resistant to the negative mating pattern and fitness changes expected within fragmented landscapes. This pattern is likely explained by strong outcrossing and regular long-distance pollen flow.
Similar content being viewed by others

Introduction
Many tree species achieve far-reaching gene flow, particularly through the movement of pollen (Petit et al., 2005; Kremer et al., 2012). For this reason, some tree species have been observed to be buffered against the expected negative population genetic effects of habitat fragmentation, which are commonly exhibited by other organisms. In this ‘paradox of forest fragmentation genetics’ (Kramer et al., 2008), high intra-population genetic diversity is usually maintained for trees, even in seemingly challenging situations (Lowe et al., 2005; Bacles and Jump, 2011; Vranckx et al., 2011).
However, within tree populations, the mating patterns of individuals (for example, inbreeding, pollen diversity) can strongly influence the gain or loss of genetic diversity over generations (Lowe et al., 2005). In fragmented tree populations, habitat loss and reduced stand density frequently disrupts pollination, which in turn alters mating patterns and gene flow dynamics (Ghazoul, 2005; Sork and Smouse, 2006). A common consequence of lower density is that altered pollination dynamics increases inbreeding (Lowe et al., 2005; Eckert et al., 2010; Breed et al., 2012) and/or reduces pollen diversity (Lowe et al., 2005; Breed et al., 2012). Nonetheless, this is not always the case (Bacles and Jump, 2011), and impacts can be highly context-dependent and affected by pollen vectors (Sork and Smouse, 2006; Eckert et al., 2010; Breed et al., 2012). To overcome this context-dependant effect, studies can investigate individuals within a single population that vary in their stand density (for example, Mimura et al., 2009; Llorens et al., 2012). This design controls for population effects that potentially influence tree responses to stand density, such as divergence in environmental (for example, pollinator community) and phenotypic (for example, mating system) traits.
Elevated inbreeding may impose a fitness cost (that is, inbreeding depression), as increased homozygosity increases the probability that the phenotypes of deleterious recessive alleles (that is, genetic load) are expressed (Crnokrak and Barrett, 2002; Szulkin et al., 2010) and is expected to be most severe in stressful environments (Fox and Reed, 2010). Trees predominantly outcross, allowing them to accumulate genetic load, and are thus likely to exhibit inbreeding depression (Crnokrak and Barrett, 2002). Statistical associations between an offspring’s heterozygosity at neutral genetic markers and its fitness are expected to occur because of changes to inbreeding rates due to disrupted pollinator behaviour. These heterozygosity–fitness correlations (HFCs) describe how variation in inbreeding (caused by nonrandom matings, small population sizes/bottlenecks or admixture/migration) associates with variation in fitness (Szulkin et al., 2010). The potentially weak association between neutral marker heterozygosity and fitness has been raised as a criticism of HFCs (Balloux et al., 2004; Pemberton, 2004; Slate et al., 2004). This issue originates from heterozygosity at neutral markers being indirectly related to fitness. However, it is still valid to observe HFCs in natural populations despite this limitation; and HFCs tend to be easier to conduct on natural populations than the alternative pedigree-style studies, which require pedigree records over several generations (Szulkin et al., 2010). HFCs are expected to be present when there is variation in inbreeding in the study population. Detection requires that many individuals are genotyped, as the HFC effect is often weak and because only few microsatellite loci tends to be genotyped. In addition, only fitness traits affected by many loci (for example, growth) are likely to be associated with heterozygosity at neutral genetic markers, as the power to detect this indirect relationship between neutral markers and observed fitness traits increases with the number of functional loci under directional selection (Szulkin et al., 2010).
Disrupted pollinator movements may reduce pollen diversity if the number or diversity of pollen sources or pollen donors decline. The commonly estimated mating-system parameter, correlated paternity, rp, describes the diversity of pollen contributions for sampled progeny arrays and is proportional to the effective number of pollen donors (Sork and Smouse, 2006). Correlated paternity is expected to be negatively correlated with fitness, as reduced pollen diversity is expected to result in a higher probability of unfit combinations of pollen and ovules via either less pollen competition or weaker heterosis (Yasui, 1998; Skogsmyr and Lankinen, 2002).
While a great deal of theoretical and empirical work has been conducted on the effects of fragmentation on tree pollination dynamics and open-pollinated progeny diversity (for example, Lowe et al., 2005; Eckert et al., 2010), few studies have investigated the downstream fitness consequences of these changed pollination dynamics. Indeed, as far as we are aware, there are currently only five published studies of open-pollinated Neotropical trees that have associated changes in mating patterns of individual trees caused by habitat disturbance with proxies of fitness (Rocha and Aguilar, 2001; Cascante et al., 2002; Fuchs et al., 2003; Quesada et al., 2004; Breed et al., 2012), and a single study conducted from Japan (Hirayama et al., 2007). As expected, selfing (4 of 6 studies), biparental inbreeding (2 of 3 studies) and correlated paternity (3 of 4 studies) tended to be negatively associated with fitness.
In this study, we combine assessments of genetic diversity, mating patterns, contemporary and historical gene flow, and proxies of progeny viability of open-pollinated progeny arrays of E. incrassata (ridge-fruited mallee). Progeny arrays were sourced from mother trees with two contrasting stand densities in a single population of fragmented woodland. E. incrassata is a multi-stemmed, predominantly bird-pollinated (Bond and Brown, 1979) tree that is a main canopy component of woodland throughout semi-arid south-eastern Australia. This study was conducted in the southern Murray Darling Basin, Australia, an area that has experienced extensive clearance (Bradshaw, 2012), with few intact E. incrassata populations. Specifically, we addressed three questions: (a) how does stand density alter the mating patterns and pollen flow of E. incrassata? (b) does the pre-clearance spatial genetic structure of E. incrassata correspond to contemporary gene flow estimates? and (c) is variation in the performance of E. incrassata progeny linked to their genetic diversity, contemporary mating patterns and/or stress caused by disease?
Materials and Methods
Study species
E. incrassata is a sclerophyllous tree, one of many Eucalyptus species common throughout the deep sand or sand-over-limestone soils of the semi-arid Murray Darling Basin (Parsons, 1969). E. incrassata generally grows from 4 to 8 m in height, with small hermaphroditic flowers, pollinated primarily by honeyeaters (Family Meliphagidae; for example, Phylidonyris novaehollandiae, New Holland honeyeater; Anthochaera carunculata, red wattlebird) and to a lesser degree, insects (Bond and Brown, 1979). E. incrassata is likely to have a mixed mating system with preferential outcrossing on the basis of observations of other Eucalyptus species (Sampson et al., 1989, 1995; Horsley and Johnson, 2007; Sampson and Byrne, 2008; Mimura et al., 2009). Eucalypt flowers are protandrous, however, the development of flowers within and between inflorescences is sequential and gradual so that flowers in male or female phase may be in close proximity, allowing geitonogamous selfing to occur (House, 1997). The serotinous fruits are held over numerous years, with drying triggering seed-release. Seeds are small (<2 mm diameter) and gravity-dispersed. Ants generally exhaust the soil seed bank, except during particularly heavy seed release, such as post fire, when ants cannot harvest all seeds (Wellington and Noble, 1985).
Seed collection and landscape contexts
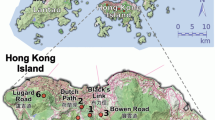
Open-pollinated seeds were collected within a single population in southern Australia from 37 mother trees across two contrasting landscape contexts: higher density intact remnant patches of woodland (n=21 trees); and lower density isolated pasture trees (n=16 trees; Figure 1). Seeds were collected from at least 10 branchlets from across the canopy of each mother tree. Branchlets generally hold many fruit (10–100 s of fruit) and each fruit contain numerous seeds (5–10 seeds per fruit; personal observations). All seeds collected from individual mother trees were mixed before sampling for the common garden experiment (see Supplementary Appendix A in the Supplementary Information for more details).

Map showing location of E. incrassata mother trees (filled circles) from both landscape contexts. Shaded areas represent native vegetation. Inset maps A, B, C give a closer view of sampled adult mother trees within the intact woodland context. All other sampled mother trees were in isolated pasture contexts. The cross ( × ) indicates the location of the common garden experiment.
Intact woodlands were natural habitats surrounded by agricultural land and trees were sampled within accessible remnant patches. Isolated pasture trees were in very small clusters of vegetation, either in agricultural land or between public roads and agricultural land. In all cases, care was taken to avoid sampling near neighbours. Stand density was characterised by (i) counting the number of adult conspecifics within a 30-m radius of the mother tree (that is, near neighbours) and extrapolating this to trees per hectare, (ii) averaging the distance to the nearest three conspecific neighbours and (iii) estimating the distance to the nearest conspecific cluster of >100 individuals within an intact remnant patch. Intact remnant woodland patches were characterised by higher density and low isolation ((i)=mean 12.6±SE 1.0 trees ha−1; (ii)=mean 35±s.e. 15 m; (iii)=0 m) with no known history of clearing. The isolated pasture trees were characterised by low density and high isolation (i=mean 1.7±s.e. 0.8 trees ha−1; ii=mean 79±s.e. 21 m; iii=1700±s.e. 347 m).
Common garden experiment
We observed fitness proxies of E. incrassata seedlings from our seed collection in a common garden experiment (n=649 seedlings), implementing a randomised complete block design (Supplementary Appendix A). All mother trees sampled were in close proximity to the planting location (<12 km) to avoid potential confounding population effects (for example, local adaptation to soil; Figure 1). Height and disease rate were the fitness proxies observed during this trial. Individual-level statistical analyses of height used raw height (hereafter height), and implemented block and family as random effects (see below for more details on statistical analyses). Family-level statistical analyses of height used block-standardised height (hereafter heightstd) methods, as described in Breed et al., (2012). To standardise the height for variation among blocks, the raw height of the ith seedling from the bth block (hib) was measured 10 months post-germination. The difference between the mean height of the bth block (ĥb) and the grand mean across all blocks (ĥ) was used to adjust hib to account for variation among blocks where adjustedhib=hib-(ĥb-ĥ)=heightstd. Using growth as a fitness proxy has limitations, as established seedlings may have experienced strong selection at pre- or early post-zygotic stages. However, germination rates were not observed to be different across groups, although detailed data were not available for further analysis. Also at this time, the presence of fungal infection of each seedling was observed. The infection was most probably Mycosphaerella sp., which causes blotchy discolouration of leaves (Keane et al., 2000), and is assumed to impose a fitness cost - hereafter ‘disease’.
Microsatellite genotyping
Leaf tissue was collected from each seedling and DNA was extracted using the Machery-Nagel Nucleospin Plant II Kit at the Australian Genome Research Facility (AGRF, Adelaide, Australia). Eight direct-labelled microsatellite markers were selected from the set of EST-derived markers by the following Faria et al., (2010; EMBRA1382; EMBRA2002; EMBRA1445; EMBRA914; EMBRA1990; EMBRA1284; EMBRA1928; EMBRA1468). A BLAST search was performed for each EST using accession numbers in Faria et al., (2010), and resulted in no significant hits with genes with a known function. PCR was performed in a single 10-μl multiplex PCR containing 1 -μl template DNA (ca. 20 ng μl−1), 5 μl 2 × Qiagen Multiplex PCR Master Mix (Qiagen, Hilden, Germany), 3 μl of water, 1 μl of primer mix with each primer at 2 μM concentration. Standard Qiagen Multiplex PCR conditions were used with an initial activation step at 95 °C for 15 min, then 40 cycles of denaturation at 94 °C for 30 s, annealing at 60 °C for 90 s, then extension at 60 °C for 60 s, with a final extension at 60 °C for 30 min. Fragments were separated on an AB3730 genetic analyser with a 36-cm capillary array (Applied Biosystems, Foster City, CA, USA) and LIZ500 size standard was added to the samples before running. Alleles were sized using GeneMapper software (Applied Biosystems) and double-checked manually.
Data analysis
The most likely maternal genotypes were determined from each progeny array using MLTR (Ritland, 2002), and used to estimate genetic diversity and spatial genetic structure of the adult cohort, which was presumed to reflect pre-clearance dynamics, as all trees sampled were estimated to be >80 years old. These data were also used to screen for null alleles in MICRO-CHECKER (Oosterhout et al., 2004). GENEPOP on the web (http://genepop.curtin.edu.au) was used to assess the tests for heterozygote deficit/excess and linkage disequilibrium, applying sequential Bonferroni’s correction for multiple testing, where appropriate. The per-locus probability of paternity exclusion (Q) and combined probability of paternity exclusion (QC) were estimated using the adult mother-tree data in GENALEX (Peakall and Smouse, 2006).
Genetic diversity
For both adult (n=37) and progeny (n=649) cohorts from each landscape context, we estimated the following population genetic parameters using GENALEX: number of alleles (A); Nei’s (1973) unbiased expected and observed heterozygosity (HE and HO, respectively); and the fixation index (F). To account for differences in sample size, we estimated rarefied mean number of alleles per locus (AR) using HP-RARE (Kalinowski, 2005). We estimated individual observed progeny multilocus heterozygosity (Hi) and scaled the measure to account for missing data by Hi=Σhij/ni, where h is a heterozygote for the ith individual at the jth locus for n successfully genotyped loci. All samples that failed amplification at more than four loci were excluded (n=28). We calculated family heterozygosity as Hf=ΣHfi/nf, where Hfi is the progeny multilocus heterozygosity for the ith individual in the fth family of sample size n.
Spatial genetic structure
Spatial genetic structure of adult mother trees (n=37) was analysed using the software SPAGeDi (Hardy and Vekemans, 2002), following the procedure described in Vekemans and Hardy (2004), on the basis of pairwise kinship coefficients between individuals, Fij (Loiselle et al., 1995). To visualise the relationship between average Fij and geographic distance, we defined distance classes following the software authors’ recommendations, where, for each distance class, >50% of all individuals were represented at least once and the coefficient of variation of the number of times each individual represented was ⩽1. Average Fij was plotted over the logarithm of the distance class. Pairwise kinship coefficients were regressed on the logarithm of pairwise distance to estimate the regression slope, b, and the significance of this slope was tested with 10 000 permutations. The strength of spatial genetic structure was then quantified by calculating the Sp-statistic after Vekemans and Hardy (2004). Sp=−b/(F1-1), where F1 is the average kinship coefficient between individuals within the first distance class (<50 m in this case) and b is the regression slope of Fij regressed on the logarithm of pairwise distance. There was no convergence of gene dispersal estimates (σ) using SPAGeDi when choosing a range of effective density estimates, from 1/2 to 1/15th of the census density of intact woodland adults (12.6 trees per hectare).
Mating system and gene flow
We estimated the following mating system parameters for mother trees from each landscape context in MLTR (Ritland, 2002): multilocus outcrossing rate (tm); single-locus outcrossing rate (ts); biparental inbreeding (tm−ts); and multilocus-correlated paternity (rp). Families were bootstrapped 1000 times to calculate the variance estimates for each parameter within each landscape context. The same mating system parameters were also estimated for each family. This was conducted in the same way except that individuals within families were bootstrapped 1000 times to calculate the variance estimates.
For trees within each landscape context (that is, higher density intact woodland and lower density isolated pasture), KINDIST (Robledo-Arnuncio et al., 2007) was used to estimate the pollen dispersal kernel scale (a) and shape (b) parameters and the average effective pollen dispersal distance (δ). KINDIST models the decay in a normalised measure of correlated paternity with increasing spatial distance between mother pairs, and is independent of the density of pollen donors (Robledo-Arnuncio et al., 2006). Correlated paternity was calculated on the basis of pairwise kinship, as the probability of paternal identity between a pair of maternal sibs with geographic distance, z, relative to the average within maternal sibship (Robledo-Arnuncio et al., 2006). This indirect method estimates contemporary pollen dispersal as a function of the genetic structure of pollen pools allowing for unequal male fecundity. The usefulness of this approach was validated by testing for a decline in pairwise-correlated paternity with increasing pairwise distance using a Spearman’s rank correlation. In all cases, the exponential-power dispersal function best fitted our data, and data within a 4000-m distance cut-off were used for model fitting. Differentiation in mother-tree pollen pools (Φft) and effective density (de) across landscape contexts was estimated with TWOGENER analysis (Smouse et al., 2001), also part of the POLDISP package. Effective number of pollen donors (Nep) was estimated by Nep=rp−1 in MLTR, as well as Nep=(2Φft)−1 in TWOGENER using the shape (a) and scale (b) exponential-power function parameters estimated in KINDIST.
Statistical analyses
We used general linear and Gaussian mixed-effect models in a maximum likelihood, multi-model inference framework in R v. 2.12.1 (R Project for Statistical Computing, http://www.r-project.org; Burnham and Andersen, 2002) to analyse relationships among genetic predictors and fitness proxies for E. incrassata. We used Akaike’s Information Criterion corrected for small sample sizes (AICc) for model selection (Burnham and Andersen, 2002).
We conducted HFCs at the individual-level. We used Gaussian mixed-effect models to evaluate the relationship between individual heterozygosity (Hi) and height of all progeny (n=649), as well as for diseased seedlings only (n=54). Block and family were treated as random effects and Hi was treated as a fixed effect (see Supplementary Appendix B for more details on statistical methods). We derived the slope (βω,Hi) and variance explained (r2ω,Hi) of these models. However, although a correlation between heterozygosity and fitness does not indicate how much variation in fitness is explained by inbreeding, inbreeding load (βω,f) and variance in fitness explained by inbreeding (r2ω,f) were also estimated methods described in (Szulkin et al., 2010).
HFCs rely on variation in inbreeding resulting in a correlation between the observed heterozygosity at the genotyped loci and the heterozygosity at functional loci (that is, correlation due to identity disequilibrium). As such, the inter-locus heterozygosity correlation (g2) was estimated in RMES (David et al., 2007). However, the observed inter-locus correlation of heterozygosity was not significantly different from zero (g2=0.003, s.d.=0.015, P=0.41). A nonsignificant g2 does not preclude the detection of HFCs from the observed data, as HFCs generally have greater statistical power than g2 (Szulkin et al., 2010). As an alternative, g2 was estimated from the independently derived selfing rate using the parent–offspring data in MLTR (s=1−tm). The selfing estimate can be implemented in equation 4 in David et al., (2007). Deriving g2 in this manner is appropriate for data sets of this kind and this method may have statistical benefits to the g2 method, as the selfing rate here is derived from data independent of observed heterozygosity. The inbreeding estimate f was derived from the MLTR selfing estimate s, where f=s/(2−s).
At the family level, we used general linear models to test for hypothesised relationships between mean heightstd of each family and four genetic predictors: multilocus outcrossing rate (tm); biparental inbreeding (tm−ts); correlated paternity (rp); and family average heterozygosity (Hf). We also used the binomial generalised linear models to evaluate the effects of Hf on proportion of diseased plants in each family (Crawley, 2007).
Results
Genetic diversity and diagnostic statistics
A total of 357 progeny from 21 adults sampled in a higher density intact woodland context and 292 progeny from 16 adults sampled in a lower density isolated pasture context were genotyped (progeny array n=5–20; mean 17.5±s.e. 0.7). A total of 85 alleles were identified in the adult cohort (summary of per locus data are presented in Supplementary Appendix C in the Supplementary Information). The combined probability of paternity exclusion if neither parent is known indicates good resolution for the genetic markers used (QC=1.00; Supplementary Appendix C). No significant null alleles were detected across all loci. No significant linkage disequilibrium was observed between pairs of loci scored in the adults after adjustment for multiple testing. A significant excess of heterozygotes was observed in the adults, contributed mostly by two loci (EMBRA2002, EMBRA1445; Table 1; Supplementary Appendix C). Overall, the number of alleles detected in progeny cohorts was equivalent to adult cohorts. However, progeny were significantly more homozygous than adults were (Table 1). Genetic diversity was comparable between adults from both landscape contexts, and similarly, there was little difference in diversity for progeny across the landscape contexts.
Spatial genetic structure
Adult mother trees, which we took as representative of pre-clearance genetic structure, displayed weak spatial genetic structure (Sp=0.005; blog= −0.005; F1=0.049; P=0.09), with individuals within 50 m of each other more likely to be closely related than by chance (Figure 2). Beyond 50 m, no significant differences were observed.

Spatial autocorrelation of genetic relatedness of adult E. incrassata mother trees. Significant autocorrelation between pairwise kinship within a distance class is indicated if mean observed pairwise kinship (filled circles±s.e.) falls outside confidence interval (CI) limits (open squares). If observed autocorrelation is above the upper 95% CI line, then individuals within that distance class were more genetically similar than expected at random, and a significant positive spatial structure is observed.
Mating system and contemporary gene flow estimates
E. incrassata was primarily outcrossed (94%), and stand density had no observed effect on multilocus outcrossing rate (tm), biparental inbreeding (tm−ts) and correlated paternity (rp; Table 2).
Within each density group, we observed negative correlations between correlated paternity and distance between maternal trees, indicating appropriate use of KINDIST to estimate pollen dispersal curves (Spearman’s rank correlation: intact woodland ρ=−0.13; P=0.07; isolated pasture ρ=−0.16; P=0.08). The exponential-power function best explained variation in the correlated paternity-by-distance relationship in both landscape contexts, indicating leptokurtic (that is, fat-tailed) pollen dispersal dynamics (Table 3). The leptokurtic pollen dispersal relationship was stronger (that is, more leptokurtic) for mothers from isolated landscape contexts, corresponding to a greater estimated mean pollen dispersal distance for isolated than intact woodland-context mothers (isolated pasture δ=1130 m, intact woodland δ=870 m).
The lower stand density group had marginally greater pollen pool structure and lower effective density (Tables 2 and 3). Effective-to-census tree density ratios indicated that a much greater proportion of trees in lower stand density contexts contributed pollen to the observed generation when compared with trees in intact woodland contexts (Table 3).
Heterozygosity–fitness correlations
At the family level, there were no associations between genetic parameters and family heightstd or disease rates (null model was best fitting model or ΔAICc to null model was <2; Supplementary Appendix B).
At the individual level for all progeny, there was a weak relationship between heterozygosity and height (ΔAICc to model not including heterozygosity=2.41; wAIC=0.77; deviance explained=1.29%; Supplementary Appendix B), corresponding to very little observed inbreeding load (Table 4). However, we observed a strong positive relationship between heterozygosity and height in diseased seedlings (ΔAICc to null model=5.41; wAIC=0.90; deviance explained=2.70%; Supplementary Appendix B). Of the total 37 families, 28 showed evidence of disease in 1–4 progeny within families. By scaling our observed positive relationship between heterozygosity and heightstd by the inter-locus correlations of heterozygosity (g2), variance in inbreeding and variance in heightstd owing to environmental or genetic effects that are not related to inbreeding (see Szulkin et al., 2010 for details), the positive relationship between heterozygosity and heightstd corresponded to a large inbreeding load and translated to an average change in height after one generation of selfing (that is, f=1/2) of −39.1 cm × 1/2=−19.55 cm (observed mean seedling height=30.66 cm). Inbreeding explained 12.3% of variation in heightstd of diseased seedlings (Table 4).
Seedlings that were diseased had no difference in heterozygosity compared with non-diseased seedlings (0.63±0.17; 0.63±0.18; diseased and non-diseased, respectively, ±one s.d.).
Discussion
Resistance to fragmentation
Contrary to predictions (Lowe et al., 2005; Eckert et al., 2010; Bacles and Jump, 2011), our study found that a population of E. incrassata in South Australia appeared to have mating patterns that were resistant to fragmentation and lower stand density observed at the scale investigated here (ca. sevenfold decrease in tree density, 12.6 vs 1.7 trees per hectare; twofold increase in nearest neighbour distances, 35 m vs 79 m). This pattern of fragmentation is typical of E. incrassata populations across southern Australia (Figure 1). The variation in stand density observed did not significantly change levels of outcrossing, biparental inbreeding, correlated paternity or genetic diversity of progeny. Consequently, it is inferred that E. incrassata pollinators (primarily honeyeaters; Bond and Brown, 1979) are highly mobile within this fragmented landscape and able to provide sufficient pollination services to maternal plants in these low-stand densities, not only in terms of moving outcrossed pollen but also in maintaining pollen diversity to isolated trees. It appears that the pollinators were not restricted by having to cross the agricultural matrix present between isolated trees (mean nearest neighbour distance=79 m; mean distance to large remnant patch 1.2 km), particularly, as mean pollination distances in lower density stands were >1 km. Crossing an agricultural matrix does not provide these bird pollinators with food or shelter, thus will impose an energetic cost. In addition, a fitness cost may be imposed to these birds, as crossing agricultural land may increase their exposure, and thus their predation risk (Ford et al., 2001). Thus, the pollinators may be making movements across the landscape by using small patches of remnant vegetation distributed along roadsides and in paddocks (Figure 1). Indeed, birdbanding studies show that New Holland honeyeaters (P. novaehollandiae) are capable of movements of tens of kilometres in short time periods (Paton, 2004). In addition, our indirectly inferred gene flow estimates suggest pollen dispersal has likely been historically high in this species, as little spatial genetic structure was observed in our limited sampling of pre-clearance adult trees, although further sampling would be required to make confident conclusions about the level and significance of spatial genetic structure (Cavers et al., 2005).
Stand density, mating system and gene flow
Our results show that E. incrassata was strongly preferentially outcrossing, with only low levels of selfing (ca. 6%). This suggests that the species may potentially have a leaky genetic self-incompatibility mechanism or other means to reduce or prevent self-fertilisation (for example, pronounced protandry, selective abortion). However, these inferences remain to be tested. The high degree of outcrossing contrasts with observations of other closely related species, where 60–80% outcrossing has generally been observed for Eucalyptus species (Sampson et al., 1989, 1995; Sampson and Byrne, 2008; Mimura et al., 2009), although self-incompatibility is expected for some Eucalyptus species on the basis of controlled pollination experiments (Horsley and Johnson, 2007).
On the basis of optimal foraging theory, pollinators are expected to spend extended periods in maternal plants in lower density stands, increasing the probability of self-pollination (Ottewell et al., 2009), which is supported by data from other tree species (Lowe et al., 2005; Sork and Smouse, 2006; Eckert et al., 2010). However, in this study, lower stand densities had no detectable effect on outcrossing rate. Similarly Ottewell et al., (2009), found no increase in selfing with increased tree isolation (up to a maximum distance to nearest neighbour of 380 m), in the primarily bird-pollinated E. leucoxylon. However, in both studies, selfed seed may have been purged before sampling either through pre- or early post-zygotic mechanisms, therefore, we may not be detecting the ‘true’ selfing rate, but rather the outcrossing rate at the established seedling stage.
Our indirect estimates of pollination distances were greater, on average, among E. incrassata maternal plants in lower density isolated pasture contexts than those located in higher density intact woodland contexts (intact woodland δ=870 m; isolated pasture δ=1133 m). In addition, maternal plants in lower density stands were approximately five times more likely to be participating in reproduction over the observed generation than trees in an intact habitat context (intact woodland de/d=0.11; isolated pasture de/d=0.55). The average pollen dispersal distances estimated here, for both intact and lower density landscape contexts, are at the upper end of the range in comparison to other studies on Eucalyptus species using similar indirect methods (remnant Eucalyptus stand: 324–493 m, Sampson and Byrne, 2008; 87–149 m, Mimura et al., 2009; 25–64 m, Field et al., 2011; fragmented Eucalyptus stand: 69–833 m, Mimura et al., 2009). An increase in average pollination distance in a lower density compared with higher density and more intact habitat context is perhaps to be expected for tree species with labile pollinators (for example, birds: Mimura et al., 2009; mobile insects: Dick, 2001; White et al., 2002; Jha and Dick, 2010). More detailed analysis of pollen flow using direct parentage analysis would certainly be a fruitful line of enquiry to better understand gene flow dynamics in the fragmented landscape we studied here, including understanding the direction of gene flow between isolated trees and fragments. When considered together with the high outcrossing nature of this population, the likely high gene flow mediated by pollinator movement offers an explanation for the observed resistance of mating patterns to low density for this species.
Biparental inbreeding can confound selfing estimates, potentially leading to selfing being detected in self-incompatible populations (Ritland, 2002; Eckert et al., 2010). This artefact may be present here as families in both landscape contexts exhibited relatively high degrees of biparental inbreeding in comparison to other eucalypts in similar contexts (that is, E. incrassata intact woodland tm−ts=0.16; isolated pasture tm−ts=0.19; compared with E. globulus continuous populations tm−ts=0.04–0.06; fragmented populations tm−ts=0.06–0.11; Mimura et al., 2009). Thus, given that nearest neighbours were more closely related than random expectations, and if pollinators mostly travel short distances (that is, between near neighbours) as expected, then regular matings are likely to occur between closely related individuals. This is likely in both landscape contexts, as although we observed a greater mean distance to nearest neighbours in lower density stands, the observed distance to nearest neighbours approximated the distance where significant spatial genetic structure was observed (significant spatial genetic structure at 50 m; intact woodland: mean distance to nearest neighbours=35 m; isolated pasture: mean distance to nearest neighbours=79 m).
Stand density and genetic diversity
We observed similar genetic diversity estimates between progeny from both intact and isolated landscape contexts and between progeny and adult cohorts in the isolated landscape context. This pattern highlights the potentially beneficial nature of maintenance of long-distance gene flow, together with preferential outcrossing, for maintenance of genetic diversity.
Heterozygosity-fitness and mating system–fitness correlations
Across both landscape contexts, the progeny cohort had higher observed homozygosity than the adult cohort. This difference was made up of two components: adults were more heterozygous than expected under Hardy–Weinberg equilibrium and progeny had lower heterozygosity than adults did. This discrepancy is potentially explained by selection favouring heterozygosity at fitness-linked loci and that the heterozygosity at our microsatellite loci was correlated with functional fitness (that is, microsatellite heterozygosity covaries with functional loci heterozygosity resulting in identity disequilibrium; Szulkin et al., 2010). If this was the case, the adult cohort is likely to have undergone selection against inbred individuals, increasing their mean observed heterozygosity. Because of the long-lived nature of this species (10 s to 100 s of years) and the episodic high-stress environment caused by, for example, water availability, extreme temperatures and disease (Parsons, 1969; Keane et al., 2000; Fox and Reed, 2010), there have likely been many opportunities within the lifespan of the standing adults for inbred adults to be selected out of the population (Costa e Silva et al., 2010).
In both landscape contexts, a range of inbred and outbred progeny were produced. All seedlings experienced warm and moist rearing and field conditions (Supplementary Appendix A), which should be amenable to low-stress growing conditions for E. incrassata (Parsons, 1969). As a result, selection against inbred progeny may not have been strong in the common garden experiment (Fox and Reed, 2010). However, we still observed preliminary evidence for stress-induced inbreeding depression (Fox and Reed, 2010), with stress caused by fungal infection (Mycosphaerella sp.; Keane et al., 2000), but this was not linked to landscape context. Variation in growth explained by heterozygosity for all seedlings and diseased-only seedlings was small (<4%, Table 4), but HFC values such as this are the norm for microsatellite studies of strong outcrossing species (Chapman et al., 2009; Szulkin et al., 2010). Furthermore, as we only genotyped eight microsatellite loci to infer the inbreeding coefficients, caution is warranted when interpreting its relationship with fitness (Balloux et al., 2004; Slate et al., 2004). Thus, before confirming our conclusions about inbreeding depression and the purging of inbred E. incrassata, follow-up studies that use more sophisticated genetic and longer-term fitness assessments need to be performed.
Conclusions
Our study demonstrates that E. incrassata displayed mating patterns (that is, genetic diversity, inbreeding, pollen diversity) and potential fitness (as measured by growth) that were resistant to severe habitat fragmentation and low tree density, a pattern that goes against expectations (Young et al., 1996; Lowe et al., 2005; Eckert et al., 2010; Bacles and Jump, 2011; Breed et al., 2012). We presented concordant evidence of likely high gene flow from historical perspectives, with isolation increasing mean pollen dispersal distances. High gene flow was mediated by bird pollinators that are likely able to move long distances over the potentially hostile matrix found between isolated trees and remnant fragments. We observed preliminary evidence for inbreeding depression related to stress caused by fungal infection, but this inbreeding depression was not associated with landscape context. Together, our results suggest that E. incrassata may be buffered against the negative genetic effects of habitat fragmentation through strong preferential outcrossing and frequent long-distance gene flow. As long as recruitment or revegetation is sufficient to overcome population declines owing to senescence, an optimistic outlook is warranted for this species within fragmented landscapes.
Data archiving
Growth, block, genotype and spatial information are available through Dryad at doi:10.5061/dryad.4k660.
References
Bacles CFE, Jump AS (2011). Taking a tree's perspective on forest fragmentation genetics. Trends Plant Sci 16: 13–18.
Balloux F, Amos W, Coulson T (2004). Does heterozygosity estimate inbreeding in real populations? Mol Ecol 13: 3021–3031.
Bond HW, Brown WL (1979). The exploitation of floral nectar in Eucalyptus incrassata by honeyeaters and honeybees. Oecologia 44: 105–111.
Bradshaw CJA (2012). Little left to lose: deforestation and forest degradation in Australia since European colonisation. J Plant Ecol 5: 109–120.
Breed MF, Gardner MG, Ottewell K, Navarro C, Lowe A (2012). Shifts in reproductive assurance strategies and inbreeding costs associated with habitat fragmentation in Central American mahogany. Ecol Lett 15: 444–452.
Burnham KP, Andersen DR (2002) Model selection and multimodel inference 2nd edn. Springer New York.
Cascante A, Quesada M, Lobo JJ, Fuchs EA (2002). Effects of dry tropical forest fragmentation on the reproductive success and genetic structure of the tree Samanea saman. Conserv Biol 16: 137–147.
Cavers S, Degen B, Caron H, Lemes MR, Margis R, Salgueiro F et al (2005). Optimal sampling strategy for estimation of spatial genetic structure in tree populations. Heredity 95: 281–289.
Chapman JR, Nakagawa S, Coltman DW, Slate J, Sheldon BC (2009). A quantitative review of heterozygosity–fitness correlations in animal populations. Mol Ecol 18: 2746–2765.
Costa e Silva J, Hardner C, Tilyard P, Pires AM, Potts BM (2010). Effects of inbreeding on population mean performance and observational variances in Eucalyptus globulus. Ann For Sci 67: 605.
Crawley M (2007) The R Book. John Wiley & Sons, Ltd Chichester, UK.
Crnokrak P, Barrett SCH (2002). Purging the genetic load: a review of the experimental evidence. Evolution 56: 2347–2358.
David P, Pujol B, Viard F, Castella V, Goudet J (2007). Reliable selfing rate estimates from imperfect population genetic data. Mol Ecol 16: 2474–2487.
Dick C (2001). Genetic rescue of remnant tropical trees by an alien pollinator. Proc R Soc Biol Sci Ser B 268: 2391–2396.
Eckert CG, Kalisz S, Geber MA, Sargent R, Elle E, Cheptou P-O et al (2010). Plant mating systems in a changing world. Trends Ecol Evol 25: 35–43.
Faria DA, Mamani EMC, Pappas MR, Pappas GJ, Grattapaglia D (2010). A selected set of EST-derived microsatellites, polymorphic and transferable across 6 species of Eucalyptus. J Hered 101: 512–520.
Field DL, Ayre DJ, Whelan RJ, Young AG (2011). The importance of pre-mating barriers and the local demographic context for contemporary mating patterns in hybrid zones of Eucalyptus aggregata and Eucalyptus rubida. Mol Ecol 20: 2367–2379.
Ford HA, Barrett GW, Saunders DA, Recher HF (2001). Why have birds in the woodlands of Southern Australia declined? Biol Conserv 97: 71–88.
Fox CW, Reed DH (2010). Inbreeding depression increases with environmental stress: an experimental study and meta-analysis. Evolution 65: 246–258.
Fuchs E, Lobo J, Quesada M (2003). Effects of forest fragmentation and flowering phenology on the reproductive success and mating patterns of the tropical dry forest tree Pachira quinata. Conserv Biol 17: 149–157.
Ghazoul J (2005). Pollen and seed dispersal among dispersed plants. Biol Rev 80: 413–443.
Hardy OJ, Vekemans X (2002). SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2: 618–620.
Hirayama K, Ishida K, Setsuko S, Tomaru N (2007). Reduced seed production, inbreeding, and pollen shortage in a small population of a threatened tree, Magnolia stellata. Biol Conserv 136: 315–323.
Horsley TN, Johnson SD (2007). Is Eucalyptus cryptically self-incompatible? Ann Bot 100: 1373–1378.
House SM (1997). Reproductive biology of eucalypts. In: Williams JE, Woinarski JCZ, (eds). Eucalypt Ecology: Individuals to Ecosystems. University Press Cambridge. pp 30–55.
Jha S, Dick CW (2010). Native bees mediate long-distance pollen dispersal in a shade coffee landscape mosaic. Proc Natl Acad Sci USA 107: 13760–13764.
Kalinowski ST (2005). HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5: 187–189.
Keane P, Kile G, Podger F, Brown B (2000) Diseases and Pathogens of Eucalypts. CSIRO Publishing Collingwood, Australia.
Kramer AT, Ison JL, Ashley MV, Howe HF (2008). The paradox of forest fragmentation genetics. Conserv Biol 22: 878–885.
Kremer A, Ronce O, Robledo-Arnuncio JJ, Guillaume F, Bohrer G, Nathan R et al (2012). Long-distance gene flow and adaptation of forest trees to rapid climate change. Ecol Lett 15: 378–392.
Llorens TM, Byrne M, Yates CJ, Nistelberger HM, Coates DJ (2012). Evaluating the influence of different aspects of habitat fragmentation on mating patterns and pollen dispersal in the bird-pollinated Banksia sphaerocarpa var. caesia. Mol Ecol 21: 314–328.
Loiselle BA, Sork VL, Nason J, Graham C (1995). Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82: 1420–1425.
Lowe AJ, Boshier D, Ward M, Bacles CFE, Navarro C (2005). Genetic resource impacts of habitat loss and degradation; reconciling empirical evidence and predicted theory for neotropical trees. Heredity 95: 255–273.
Mimura M, Barbour RC, Potts BM, Vaillancourt RE, Watanabe KN (2009). Comparison of contemporary mating patterns in continuous and fragmented Eucalyptus globulus native forests. Mol Ecol 18: 4180–4192.
Nei M (1973). Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70: 3321–3323.
Oosterhout CV, Hutchinson WF, Wills DPM, Shipley P (2004). MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538.
Ottewell KM, Donnellan SC, Lowe AJ, Paton DC (2009). Predicting reproductive success of insect- versus bird-pollinated scattered trees in agricultural landscapes. Biol Conserv 142: 888–898.
Parsons RF (1969). Physiological and ecological tolerances of Eucalyptus incrassata and E. socialis to edaphic factors. Ecology 50: 386–390.
Paton D (2004). Birdscaping the environment: restoring the woodland systems of the Mt Lofty region, South Australia. In: Lunney D, (ed) Conservation of Australia’s Forest Fauna 2nd edn. Royal Zoological Society of New South Wales: Mosman NSW, Australia.
Peakall R, Smouse PE (2006). GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6: 288–295.
Pemberton JM (2004). Measuring inbreeding depression in the wild: the old ways are the best. Trends Ecol Evol 19: 613–615.
Petit RJ, Duminil J, Fineschi S, Hampe A, Salvini D, Vendramin GG (2005). Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol Ecol 14: 689–701.
Quesada M, Stoner KE, Lobo JA, Herrerias-Diego Y, Palacios-Guevara C, Munguía-Rosas MA et al (2004). Effects of forest fragmentation on pollinator activity and consequences for plant reproductive success and mating patterns in bat-pollinated Bombacaceous trees. Biotropica 36: 131–138.
Ritland K (2002). Extensions of models for the estimation of mating systems using n independent loci. Heredity 88: 221–228.
Robledo-Arnuncio JJ, Austerlitz F, Smouse PE (2006). A new method of estimating the pollen dispersal curve, independently of effective density. Genetics 173: 1033–1045.
Robledo-Arnuncio JJ, Austerlitz F, Smouse PE (2007). POLDISP: a software package for indirect estimation of contemporary pollen dispersal. Mol Ecol Notes 7: 763–766.
Rocha OJ, Aguilar G (2001). Reproductive biology of the dry forest tree Enterolobium cyclocarpum (guanacaste) in Costa Rica: a comparison between trees left in pastures and trees in continuous forest. Am J Bot 88: 1607–1614.
Sampson J, Hopper S, James S (1995). The mating system and genetic diversity of the australian arid zone mallee, Eucalyptus rameliana. Aust J Bot 43: 461–474.
Sampson JF, Byrne M (2008). Outcrossing between an agroforestry plantation and remnant native populations of Eucalyptus loxophleba. Mol Ecol 17: 2769–2781.
Sampson JF, Hopper SD, James SH (1989). The mating system and population genetic structure in a bird-pollinated mallee, Eucalyptus rhodantha. Heredity 63: 383–393.
Skogsmyr IO, Lankinen Å (2002). Sexual selection: an evolutionary force in plants? Biol Rev 77: 537–562.
Slate J, David P, Dodds KG, Veenvliet BA, Glass BC, Broad TE et al (2004). Understanding the relationship between the inbreeding coefficient and multilocus heterozygosity: theoretical expectations and empirical data. Heredity 93: 255–265.
Smouse PE, Dyer RJ, Westfall RD, Sork VL (2001). Two-generation analysis of pollen flow across a landscape. I. Male gamete heterogeneity among females. Evolution 55: 260–271.
Sork VL, Smouse PE (2006). Genetic analysis of landscape connectivity in tree populations. Landsc Ecol 21: 821–836.
Szulkin M, Bierne N, David P (2010). Heterozygosity-fitness correlations: a time for reappraisal. Evolution 64: 1202–1217.
Vekemans X, Hardy OJ (2004). New insights from fine-scale spatial genetic structure analyses in plant populations. Mol Ecol 13: 921–935.
Vranckx GUY, Jacquemyn H, Muys B, Honnay O (2011). Meta-analysis of susceptibility of woody plants to loss of genetic diversity through habitat fragmentation. Conserv Biol 26: 228–237.
Wellington AB, Noble IR (1985). Post-fire recruitment and mortality in a population of the mallee Eucalyptus incrassata in semi-arid, south-eastern Australia. J Ecol 73: 645–656.
White GM, Boshier DH, Powell W (2002). Increased pollen flow counteracts fragmentation in a tropical dry forest: an example from Swietenia humilis Zuccarini. Proc Natl Acad Sci USA 99: 2038–2042.
Yasui Y (1998). The ‘genetic benefits’ of female multiple mating reconsidered. Trends Ecol Evol 13: 246–250.
Young A, Boyle T, Brown T (1996). The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11: 413–418.
Acknowledgements
This work was supported by the Australian Research Council Linkage project (LP110200805) and the South Australian Premier's Science and Research Fund awarded to AJL, funding from the Native Vegetation Council of South Australia (grant 09/10/27), Nature Foundation SA Inc., Australian Geographic Society, Biological Society of South Australia, Field Naturalist Society of South Australia, Wildlife Preservation Society of Australia and National Climate Change Adaptation Research Facility Travel Grants awarded to MFB. We would like to thank Patrice David for discussions on HFCs, Juan Jose Robledo-Arnuncio for discussions on POLDISP, Barbara Hall for assistance identifying the fungal disease, Mt Lofty Botanic Gardens staff for assistance in rearing the seedlings and Rob Murphy and the many volunteers for assistance with the common garden experiment.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Heredity website
Rights and permissions
About this article
Cite this article
Breed, M., Ottewell, K., Gardner, M. et al. Mating system and early viability resistance to habitat fragmentation in a bird-pollinated eucalypt. Heredity 115, 100–107 (2015). https://doi.org/10.1038/hdy.2012.72
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2012.72
Keywords
This article is cited by
-
Genetic assessment of the value of restoration planting within an endangered eucalypt woodland
Scientific Reports (2023)
-
Reproductive biology and population structure of the endangered shrub Grevillea bedggoodiana (Proteaceae)
Conservation Genetics (2023)
-
Gene flow, mating patterns and inbreeding depression in Roupala montana var. brasiliensis, a neotropical timber species
New Forests (2023)
-
Unexpectedly low paternal diversity is associated with infrequent pollinator visitation for a bird-pollinated plant
Oecologia (2021)
-
Spatial variation in bird pollination and its mitigating effects on the genetic diversity of pollen pools accepted by Camellia japonica trees within a population at a landscape level
Heredity (2020)
