
Abstract
Gene flow (defined as allele exchange between populations) and gene flux (defined as allele exchange during meiosis in heterokaryotypic females) are important factors decreasing genetic differentiation between populations and inversions. Many chromosomal inversions are under strong selection and their role in recombination reduction enhances the maintenance of their genetic distinctness. Here we analyze levels and patterns of nucleotide diversity, selection and demographic history, using 37 individuals of Drosophila subobscura from Mount Parnes (Greece) and Barcelona (Spain). Our sampling focused on two frequent O-chromosome arrangements that differ by two overlapping inversions (OST and O3+4), which are differentially adapted to the environment as observed by their opposing latitudinal clines in inversion frequencies. The six analyzed genes (Pif1A, Abi, Sqd, Yrt, Atpα and Fmr1) were selected for their location across the O-chromosome and their implication in thermal adaptation. Despite the extensive gene flux detected outside the inverted region, significant genetic differentiation between both arrangements was found inside it. However, high levels of gene flow were detected for all six genes when comparing the same arrangement among populations. These results suggest that the adaptive value of inversions is maintained, regardless of the lack of genetic differentiation within arrangements from different populations, and thus favors the Local Adaptation hypothesis over the Coadapted Genome hypothesis as the basis of the selection acting on inversions in these populations.
Similar content being viewed by others

Introduction
Chromosomal inversions are a major mechanism shaping the level and distribution of genomic diversity within and between species. Inversion polymorphism has been described in several Drosophila species (Krimbas and Powell, 1992) and has been observed to vary seasonally, and with altitude and latitude (Dobzhansky, 1943; Krimbas, 1993; Kennington et al., 2006). The rich polymorphism in paracentric inversions found in Drosophila subobscura (Krimbas and Powell, 1992; Krimbas, 1993) has had an important role in demonstrating the adaptive role of inversions, as shown by the coinciding latitudinal clines on inversion frequencies found in Palearctic and also in colonizing populations (Krimbas and Loukas, 1980; Prevosti et al., 1988; Balanyà et al., 2003). Moreover, changes in chromosomal polymorphism matching global warming (Solé et al., 2002; Balanyà et al., 2004, 2006) also suggest the adaptive value of inversions in D. subobscura. Despite the evidence for an adaptive role of inversions, the genetic and mechanistic basis underlying their role in adaptation remains unknown.
Several hypotheses have been suggested to explain the maintenance of inversion polymorphism in populations (reviewed in Hoffmann and Rieseberg, 2008). The coadaptation hypothesis (Dobzhansky, 1950) suggests that natural selection maintains favorable combinations of alleles that interact epistatically within and between arrangements in a certain population. The local adaptation hypothesis predicts that inversions will spread in a population at migration-selection balance when they capture at least two advantageous alleles that are individually adapted to local conditions (Kirkpatrick and Barton, 2006). Particularly, it explains that the inversion will establish a cline when interacting populations are distributed along an environmental gradient. In both hypotheses, the reduced recombination between inversion heterokaryotypes avoids gene exchange with other genetic backgrounds, but epistasis is only a requirement in the former one.
The low gene transfer between inversions (gene flux) for genes located inside the inverted region observed in some Drosophila species is in agreement with both hypotheses (Laayouni et al., 2003; Schaeffer et al., 2003; Munté et al., 2005; Hoffmann and Rieseberg, 2008). However, despite the fact that Dobzhansky (1950) detected a lower fitness of heterozygous individuals from different populations of D. pseudoobscura in laboratory experiments, molecular studies failed to detect genetic differentiation within inversions sampled from different populations (Schaeffer et al., 2003). In D. subobscura, high genetic differentiation between European populations was detected when chromosomal arrangements were used as markers, as their frequency widely varies between populations (Krimbas, 1993). However, given that these chromosomal arrangements are under strong selection (Prevosti et al., 1988; Balanyà et al., 2006), gene flow between populations would likely be underestimated using the inversions themselves as makers. Interestingly, low levels of genetic differentiation between European populations of D. subobscura were observed using molecular markers such as restriction fragment-length polymorphisms (Rozas et al., 1995) and microsatellite loci (Pascual et al., 2001). Consequently, gene flow and gene flux could be changing the genetic content of inversions from widely separated populations. Thus, the analysis of candidate genes undergoing selection in D. subobscura could allow contrasting different hypotheses explaining the maintenance of inversion polymorphism in populations.
In the present study we analyze DNA sequence variation and differentiation at six genes across the O-chromosome within and between two populations of D. subobscura from the western and eastern mediterranean: Barcelona (Spain) and Mount Parnes (Greece). Despite being located at approximately the same latitude, these populations differ substantially in inversion frequencies (Krimbas and Loukas, 1980; Krimbas, 1993; Araúz et al., 2009). Our sampling focused on homokaryotypic individuals carrying either of two arrangements, OST and O3+4, selected because they are the more common arrangements in both populations and yet show significant latitudinal clines of opposite sign (Prevosti et al., 1988). Furthermore, differential basal expression of the Hsp70 gene, a candidate for thermal adaptation, was detected between carriers of these two arrangements from the same population (Calabria et al., 2012). Thus, the six nuclear genes studied in the present work (Table 1) were chosen, because they are candidates to thermal adaptation (Laayouni et al., 2007) and because of their cytological location across the O-chromosome (Figure 1).

Top: location of the six gene regions studied in the O3+4 and OST arrangements in comparison with the ancestral O3 arrangement. The O-chromosome is divided in sections SI and SII, where SI is the fragment covered by the overlapped inversions O3+4. Bottom: sequenced fragment for each locus. Exonic regions are identified using gray boxes, and intronic regions are represented by lines. For each gene, the size of the sequenced fragment is given in parentheses.
Materials and methods
Fly samples, DNA isolation and sequencing
D. subobscura isochromosomal lines were established from two distant European populations (Barcelona, Spain, and Mt. Parnes, Greece) following the pattern of genetic crosses described in Mestres et al. (1998). Chromosomal arrangements for each line were determined by analysis of polytene chromosomes of third instar larvae (Figure 2). A total of 25 O3+4 lines (13 from Barcelona and 12 from Mt. Parnes) and 12 OST lines (5 from Barcelona and 7 from Mt. Parnes) were non-lethal and sequenced in the present study. The structure of OST and O3+4 arrangements is represented in Figure 1. It is worth considering that O3 and O4 inversions are never found alone in natural populations. D. madeirensis and D. pseudoobscura were used for interspecific analyses. D. madeirensis sequences were obtained from a laboratory strain (Supplementary Table S1) and D. pseudoobscura sequences were downloaded from Flybase (http://flybase.org).

Polytene chromosome of third instar larvae preparation of O3+4/OST heterokaryotype. The two overlapped inversions (right of the image) are located opposite to the centromere, in region SI. A full color version of this figure is available at the Heredity journal online.
Six genes distributed across the O-chromosome were studied (Figure 1): Pif1A (PFTAIRE-interacting factor 1A), Abi (Abelson interacting protein), Sqd (Squid), Yrt (Yurt), Atpα (Na pump α-subunit) and Fmr1 (Fragile X mental retardation). They were selected because they showed differential expression between laboratory populations maintained at different temperature regimes (Laayouni et al., 2007). Although their cytological location had been previously reported, new in situ hybridizations have been carried out in the present study, following standard protocols (Laayouni et al., 2000). After thorough inspection, four out of six genes proved to be previously mislocalized and the correct locations are given in Table 1. Primers used for amplification and sequencing reaction are also listed in Table 1. Some primers were reported in Laayouni et al. (2007) and others were designed in the present study using Primer Designer v1.01 (1990 Scientific and Educational Software, Durham, NC, USA). Single fly genomic DNA was extracted using Gentra Puregene Cell Kit (Qiagen, Chatsworth, CA, USA).
PCR amplifications were carried out in a 25-μl total volume with 3.5 μl buffer 10 × , 3 μl deoxyribonucleotide triphosphates (1 mM), 0.2 μl Taq DNA Polymerase (Qiagen), 0.5 μl forward primer (10 μM), 0.5 μl reverse primer (10 μM), 16.3 μl water and 1 μl DNA. Amplification conditions were 4 min at 94 °C of initial denaturation, and 35 cycles with 30 s at 94 °C, 30 s at the required annealing temperature (56 °C for all genes with the exception of Abi and Sqd at 50 °C) and 3 min at 72 °C, and 5 min at 72 °C of final extension. PCR amplification products were purified with ExoI-SAP (New England, BioLabs, Ipswich, MA, USA-Promega, Madison, WI, USA). Sequencing reactions were carried out using the ABI Prism BigDye Terminators 3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA, USA), and sequences read on an ABI 3730 sequencer at the Biotechnology Resource Center of Cornell University. Follow-up PCR and sequencing of some genes was done at the University of Barcelona as follows. PCR amplification reactions were carried out in a 15-μl total volume with 3 μl buffer 10 × , 3 μl deoxyribonucleotide triphosphates (1 mM), 0.45 μl dimethyl sulfoxide, 0.3 μl Phire Hot Start DNA Polymerase (Finnzymes, Thermo Scientific, Waltham, MA, USA), 0.75 μl forward primer (10 μM), 0.75 μl reverse primer (10 μM), 5.75 μl water and 1 μl DNA. Amplification conditions were 30 s at 98 °C of initial denaturation, and 35 cycles with 5 s at 98 °C denaturation, 5 s at the required annealing temperature (56 °C for all genes with the exception of Abi and Sqd at 50 °C) and 90 s at 72 °C, and 7 min at 72 °C for final extension. PCR amplification products were purified and sequenced as before, but at the Serveis Cientifico-Tècnics at Universitat de Barcelona. Sequences were assembled with SeqMan II (DNASTAR) and aligned with Clustal W (Thompson et al., 1994) implemented in BioEdit v7 (Hall, 1999).
Data analysis
The six analyzed genes can be divided into two groups according to their cytological location (Figure 1). The Fmr1 gene is located within inversion O4 in region SI, which includes the chromosome segment covered by the O3+4 arrangement. The other genes (Pif1A, Abi, Sqd, Yrt and Atpα) are located in region SII, which includes the colinear chromosome segment that can probably freely recombine between arrangements OST and O3+4 (Pegueroles et al., 2010a). Distance in base pairs of each gene to the nearest inversion breakpoints was estimated, assuming that all cytological bands contain the same amount of DNA and that the length of the O-chromosome of D. subobscura is equivalent to chromosome 2 of D. pseudoobscura (Pegueroles et al., 2010b). The exonic and intronic regions of each gene sequence were determined by blasting against the genome of D. pseudoobscura, and the resulting expected proteins were compared with the corresponding D. pseudoobscura proteins to ensure the correct assignment of the exons. Exonic and intronic composition of each gene is shown in Figure 1.
Analyses were carried out separately for each gene and also for the concatenated data set. The Abi gene was not included in the final concatenate data set because of the insufficient number of individuals. For this gene, sequencing of some individuals failed because of the presence of a gene duplication (data not shown). Only individuals with sequences for the five remaining genes were used in the concatenation. Gene concatenation was carried out using the Concatenator v1 software (Pina-Martins and Paulo, 2008). DnaSP v5 (Librado and Rozas, 2009) was used to estimate the standard parameters for each population and arrangement.
We estimated the number of haplotypes (h), number of polymorphic sites (S), the number of singletons, nucleotide diversity (π; Nei, 1987), nucleotide diversity in synonymous sites and non-coding positions (πsil; Nei and Gojobori, 1986) and heterozygosity in silent sites (θsil; Watterson, 1975). Comparisons in diversity values were assessed by Wilcoxon matched pairs tests using Statistica v6. Divergence per silent site (Ksil) between D. subobscura and D. madeirensis, or D. pseudoobscura (Nei and Gojobori, 1986), was also obtained. Genetic differentiation was assessed using Dxy (Nei, 1987), FST (Hudson et al., 1992) and Snn (Hudson, 2000), and its significance estimated after 10 000 replicates. These analyses were carried out excluding insertion–deletion events (indels).
Tajima’s D (Tajima, 1989) and Fu and Li’s D (Fu and Li, 1993) were used to test for neutrality. Fu and Li’s D was computed using D. pseudoobscura as an outgroup, given the similar θsil and Ksil values considering D. madeirensis as the outgroup (see results). Population size changes were determined by the statistic R2 (Ramos-Onsins and Rozas, 2002) and its significance was assessed by coalescent simulations using 1000 replicates considering either no recombination, free recombination, or level of recombination (ρ) determined from the observed sequence diversity using the composite likelihood method of Hudson (Hudson, 2001) implemented in LDhat v2.1 (http://www.stats.ox.ac.uk/~mcvean/LDhat). Gene genealogies were constructed using the neighbor-joining method with the maximum composite likelihood model implemented in the Mega v4 program (Tamura et al., 2007). Bootstrap values were obtained from 1000 replicates.
The age of inversions was estimated with the Fmr1 gene, as it is located inside the inversions, using two different approaches: using the silent nucleotide diversity of the two most divergent individuals (Rozas and Aguadé, 1994), and using mean silent nucleotide diversity of all individuals (Rozas et al., 1999), except those identified as recombinants. The number of substitutions per site and year was estimated using the divergence per silent site between D. subobscura and D. pseudoosbcura, assuming that the two species diverged 17.7 million years (Myr) ago, based on a large multilocus data set (Tamura et al., 2004), and 8 Myr ago, based on only one gene (Ramos-Onsins et al., 1998) but used for comparison with previous studies.
Results
Nucleotide variation
Genetic variability levels of the six genes within each arrangement (O3+4 and OST) and for each population are presented in Table 2. In all cases, the number of haplotypes detected approached the number of sequences analyzed. No significant differences between populations were obtained neither in nucleotide diversity (π: Z=0.58, P=0.56) nor in the number of polymorphic sites (S: Z=0.04, P=0.97). When comparing the two arrangements within populations, no significant differences were detected for π (Z=0.31, P=0.75 for Barcelona population and Z=1.36, P=0.17 for Mt. Parnes population). The number of polymorphic sites within each arrangement was similar for all genes, except for Pif1A. This gene presented the highest nucleotide diversity, probably because of its larger proportion of intronic regions (Figure 1). Thus, nucleotide diversity, heterozygosity per site and divergence were subsequently analyzed using only synonymous sites and non-coding positions. The mean highest πsil and Ksil values found in the Yrt gene (πsil=0.036, Ksil-mad=0.078, Ksil-pseu=0.325) suggest that this gene might have a higher substitution rate. Genetic divergence from D. subobscura was always higher for D. pseudoobscura than for D. madeirensis, as expected from their phylogenetic relationships (Table 2).
Genetic differentiation between populations and arrangements
When comparing populations for the same arrangement, the Snn values were always not significantly different from zero (Table 3). Between arrangements, significant differences and fixed mutations were only found for Fmr1 (Supplementary Table S2). This result is consistent with the cytological location of Fmr1 within inversion O4 (Figure 1). FST values were small for all the comparisons, with the exception of Fmr1 when comparing the two arrangements. Moreover, there were significant differences between arrangements for the concatenated data set considering all genes, but the analysis of the concatenated data set excluding Fmr1 showed small and non-significant values (Table 3). The highest Dxy values were detected for Pif1A, (Table 3), which could be explained by its large portion of intronic content resulting in higher variability levels.
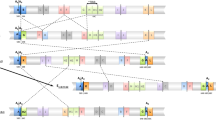
Distance trees for the concatenated data set or only for Fmr1 were carried out using D. madeirensis and D. pseudoobscura as outgroups (Figure 3). In both phylogenetic trees, sequences from different populations randomly clustered. In the tree of the concatenated data set, both populations and arrangements were randomly distributed (Figure 3a), and although some sequences from the same arrangements were grouped together, their bootstrap values were below 30%. Nonetheless, it is worth noting that in the Fmr1 genealogy (Figure 3b), the O3+4 sequences grouped with a high bootstrap value (97%), though the clustering of the remaining sequences (all belonging to the OST arrangement) was not statistically supported. By visual inspection of the Fmr1 sequences (Supplementary Table S1) we detected four recombinant sequences for the OST arrangement (asterisks show the recombinant individuals in Figure 3b), but none was detected for the O3+4 arrangement. The regions affected by recombination (more likely gene conversion due to their short size) were approximately 161 bp for MP19 and FBC58, and 10 bp for MP28 and BC55 individuals. When excluding these recombinant individuals, nine fixed differences were observed between both arrangements combining sequences from both populations (Supplementary Table S1). Furthermore, the phylogenetic reconstruction after excluding these recombinants showed monophyly for the two arrangements with more than 89% bootstrap support.

Neighbor-joining gene genealogy based on total nucleotide variation in (a) the concatenated data set of the five genes (Pif1A, Sqd, Yrt, Atpα and Fmr1) and in (b) the Fmr1 gene alone. D. madeirensis and D. pseudoobscura were used as outgroups. Only bootstrap values above 70% are shown. (Δ) O3+4 from Mt. Parnes, (▵) O3+4 from Barcelona, (◊) OST from Mt. Parnes and (♦) OST from Barcelona. Asterisks indicate recombinant individuals.
Population demography and selection
Tajima’s D and Fu and Li’s D test presented negative, although non-significant, values for the concatenated data set as well as for each gene separately, with the exception of the Atpα gene in Mt. Parnes (Table 4). Negative D statistic values correspond to an excess of low-frequency polymorphism, which could be due to either demography or selection. To further elucidate this issue, the R2 statistic (Ramos-Onsins and Rozas, 2002), which is a test for recent population expansion, was estimated because it is more suitable for small sample sizes presenting recombination. R2 estimates ranged between 0.09 and 0.18, and significance was assayed considering three alternative recombination scenarios, as rejection of the null hypothesis of constant population size depends on the level of recombination implemented (Table 4). Most of the comparisons were non-significant assuming no recombination, although they were significant when incorporating for each gene the rate of recombination (ρ) estimated using the composite likelihood method of Hudson (2001). Very similar results were obtained assuming free recombination. As the fraction of coding region varies among genes, to compare estimates previously described tests were also calculated using only silent sites (Supplementary Table S3). In general, the same patterns were detected when comparing different recombination levels with the number of significant R2 estimates being always higher in Mt. Parnes than in Barcelona. Finally, when considering population growth within arrangements for gene Fmr1 gene, which is the only gene showing signals of significant genetic differentiation, the null hypothesis of constant size was only rejected for the O3+4 arrangement (Table 4).
Age of inversions
Sequences from Fmr1 were used for estimating the age of the inversions, as it is the only gene analyzed, which is located in section SI, in which both arrangements differ (Figure 1). Recombinant individuals were excluded because, only a variation originated by mutation is useful for this analysis (Figure 3). The ages of inversions were estimated for each population separately and combining them, as their origin should be unique (Table 5). As older divergence times lead to estimate smaller mutation rates, the ages of the inversions estimated using Tamura et al. (2004) divergence time are sensitively older than using Ramos-Onsins et al. (1998) divergence time (Table 5). In addition, older estimates were obtained using the information from the two more distant individuals, in relation to the values obtained using the mean silent nucleotide diversity. Within each approach, the age estimates for O3+4 and OST were quite similar. Nonetheless, older coalescent times were detected for O3+4 when using the most divergent individuals (1.01 and 2.24 Myr for both divergence times when mixing populations, and 0.87 and 1.93 Myr for Barcelona population; see Table 5), whereas older coalescent time were detected for OST with the mean nucleotide diversity excluding recombinants (0.49 and 1.09 Myr for both divergence times when mixing populations, and 0.48 and 1.06 Myr for Barcelona population; see Table 5).
Discussion
Patterns of nucleotide variation and demographic effects
The present study focuses on the analysis of six genes in the context of two different arrangements: the OST and the O3+4. Five of these genes are located outside the inverted region and only one (Fmr1) was located inside the O4 inversion and approximately 1.3 Mb from the nearest breakpoint. In terms of nucleotide variability, lower nucleotide diversity values are expected in markers located close to the inversion breakpoint (Andolfatto et al., 2001, Stevison et al., 2011). In D. buzzatii, a correlation between nucleotide diversity and distance to the breakpoint was observed (Laayouni et al., 2003). However, Fmr1 does not present the lowest levels of πsil, despite being located inside and close to the inversion breakpoint. Similarly, no reduction of the variation was observed in D. subobscura for genes located close to the inversion breakpoints (Munté et al., 2005). Mutation and recombination (including gene conversion) could both have contributed to recover variability levels within the inverted regions. In fact, the variability for Fmr1 gene in OST was increased because of the presence of four putative recombinant individuals, despite this gene is closely located to O4 inversion breakpoint. The presence of recombinants only among OST lines could arise from differences in the frequency of both arrangements in the two populations. According to data from Araúz et al. (2009), in Barcelona and Mt. Parnes it is more likely to find O3+4+X homozygous (35% and 74%, respectively) than OST/O3+4+X heterozygous (13% and 18%, respectively) or OST homozygous (1.3% and 1.1%, respectively), where X includes non-overlapped inversions that can freely recombine in region SI with O3+4 (Figure 1). Therefore, the probability of sampling a recombinant OST chromosome is higher than that for a recombinant O3+4+X. Similarly, a previous study concluded that the frequency of heterozygote hybrids between D. pseudoobscura and D. persimilis determines the frequency of genetic exchange in the population (Stevison et al., 2011).
It has been previously suggested that recombination should be higher in central parts of the inversion, because genes located in this region could be influenced by double crossovers and gene conversion, whereas genes close to inversions breakpoints should be mainly influenced by gene conversion (Navarro et al., 1997). Nevertheless, double recombinants have been observed close to the breakpoints when analyzing the offspring of heterokaryotypic females, indicating that recombination close to the breakpoint can occur, although in very low frequency (Pegueroles et al., 2010a). As recombination between heterokaryotypes is reduced inside inverted regions (Navarro et al., 1997; Stump et al., 2007, Pegueroles et al., 2010a), higher diversity levels are expected outside inverted regions than inside them. In the region where genes can freely recombine between both arrangements (SII region, Figure 1), estimates of πsil ranged from 0.004 to 0.024 for the OST arrangement and from 0.006 to 0.023 for O3+4, after excluding Yrt, as it could act as an outlier for the nucleotide variability (see Results section). The πsil values for nine genes located inside the inverted region between the two assayed arrangements (SI region, Figure 1), combining results of a previous study (Munté et al., 2005) and the present work, ranged from 0.004 to 0.013 for the OST arrangement, and from 0.008 to 0.013 for the O3+4 arrangement, after excluding the Acph-1 gene, which also acted as a mutation outlier. The ranges of nucleotide variability mostly overlap for genes located inside and outside the inverted regions, and non-significant differences were detected when performing a Mann–Whitney U-test (OST P=0.142, O3+4 P=0.457), although the highest values were found outside the inversions. Reduced diversity within inversions has been found in the D. persimilis lineage, indicative of a recent fixation process, despite inversions may have arisen long ago (McGaugh and Noor, 2012). Our results suggest that despite the studied inversions are maintained by selection (Prevosti et al., 1988, Balanyà et al., 2006), recombination could also contribute in some extent to recover variability inside reasonably old polymorphic inversions.
The trend in the excess of rare alleles detected by the Tajima’s D in Barcelona and Mt. Parnes populations is similar to that observed for genes located inside the inverted regions in previous studies (Rozas et al., 1999; Munté et al., 2005). The hypothesis of a recent population expansion was further supported by the R2 statistic. The number of significant R2 estimates is higher in Mt. Parnes than in Barcelona, suggesting that Mt. Parnes had a larger population expansion in accordance with its estimated larger effective population size (Araúz et al., 2009, 2011). When considering expansion within arrangements, the null hypothesis of constant size was only rejected for O3+4 arrangement. As this arrangement is considered to be warm adapted (Balanyà et al., 2004), the inferred expansion in the O3+4 arrangement could be the result of its increase in frequency because of selection after the Pleistocenic glaciations. Interestingly, this arrangement shows increased frequency worldwide, matching recent global warming (Balanyà et al., 2006).
Divergence time of inversions
The age of inversions can be estimated considering their likely unique origin (Powell, 1997). These estimates can be inferred from the time of coalescence of the sequences, taking into account that variability accumulated in the sequences is proportional to its origin. Divergence was initially estimated using two species of the obscura group as outgroups (D. madeirensis and D. pseudoobscura). On one hand, D. madeirensis is more closely related to D. subobscura, as their divergence time was estimated at about 0.63 Myr ago by Ramos-Onsins et al. (1998). This species is endemic of Madeira Island and has a small effective population size. However, its nucleotide diversity is similar to D. subobscura, and therefore not showing the lower levels of variation typically found in an island (Ramos-Onsins et al., 1998; Khadem et al., 2001). On the other hand, D. pseudoobscura divergence time with respect to D. subobscura was estimated to be 17.7 Myr (Tamura et al., 2004) or around 8 Myr (Ramos-Onsins et al., 1998). D. pseudoobscura is a native species from North America and its effective size could be quite similar to D. subobscura (Pascual et al., 2000). For the Abi and Atpα genes, values of silent nucleotide diversity were quite similar to the divergence between D. subobscura and D. madeirensis. Thus, we have used the more divergent outgroup, D. pseudoobscura, to estimate the age of the inversions.
Ages were calculated using two divergence times between D. subobscura and D. pseudoobscura. The divergence time obtained by Tamura et al. (2004) is more reliable, as it is based on a large multilocus data set; nonetheless, the time estimate by Ramos-Onsins et al. (1998), although only based on the rp49 gene, allows the comparison with previous studies (see below). The age of inversions estimated using the two most divergent sequences seems more sensitive to differences in sample size or to differences in the genetic content between populations, as observed by the higher fluctuation of the estimated ages when mixing populations (1.01–2.24 Myr) or estimating ages for each population separately (0.77–1.70 and 0.87–1.93 Myr for Barcelona and Mt. Parnes, respectively, see Table 5). To avoid this potential bias, we suggest using the mean πsil and considering individuals from a unique population (bold values in Table 5). The age estimates obtained in the present study (0.48 Myr for OST and 0.41 Myr for O3+4) are slightly older than those based on genes Acph-1 (0.26 Myr for OST and 0.31 Myr for O3+4 in Navarro-Sabaté et al., 1999) and rp49 (0.24 Myr for OST and 0.33 Myr for O3+4 in Rozas et al., 1999), despite using the same divergence time (Ramos-Onsins et al., 1998). Differences could be due to intrinsic characteristics of the different genes and to their distinct genetic location. The genes Acph-1 and rp49 are closely located inside inversion O3 near its distal breakpoint (Munté et al., 2005), whereas the Fmr1 gene is located far away from them, inside inversion O4 near the distal breakpoint. However, in the three studies the age distance between the two arrangements is reduced, differing in 0.05 Myr in (Navarro-Sabaté et al., 1999), in 0.09 Myr in (Rozas et al., 1999), and in 0.07 Myr in the present study. As genes can differ in their selective pressure, mutation rates, or rate of recombination, each of which can affect estimates of their coalescence time, a multilocus approach as in Tamura et al. (2004) is preferred to more precisely infer the age of inversions. Thus, considering all data available we can conclude that the two arrangements arose approximately at the same time.
Gene flux is not homogeneously distributed across the chromosomal arrangement
Gene flux is defined as the probability of allele exchange during meiosis in heterokaryotypic females, including both crossing over and gene conversion (Navarro et al., 1997). For the five genes located outside the inversions, no significant genetic differentiation between the OST and O3+4 chromosomal arrangements was observed in the present study, suggesting extensive gene flux between them. High levels of gene flux between arrangements were also found in other Drosophila species, such as D. pseudoobscura (Schaeffer and Anderson, 2005) and D. buzzatii (Laayouni et al., 2003). Despite the extensive exchange found outside the inverted region, significant genetic differentiation was found within the inverted region. For the Fmr1 gene, the FST values were similar to those obtained in previous studies using other genes located within the studied inverted region (Rozas et al., 1999; Munté et al., 2005). The adaptive value of inversions in D. subobscura has been supported by many observations, such as the latitudinal clines for some chromosomal arrangements (Prevosti et al., 1988; Balanyà et al., 2006) or their seasonal fluctuations (Rodríguez-Trelles et al., 1996). In particular, OST and O3+4 present opposite latitudinal clines, with OST being more frequent in northern Europe and O3+4 being more frequent in the south (Solé et al., 2002; Balanyà et al., 2004). However, none of the nine genes located across the O3+4 arrangement studied so far show any non-synonymous changes differentiating the two arrangements (Munté et al., 2005 and present study), despite the Fmr1 gene was a candidate gene to be involved in thermal adaptation. Knowing that selective pressure could focus on regulatory regions instead of coding regions (Torgerson et al., 2009), the differential basal expression for the thermal candidate gene Hsp70 among O3+4 and OST arrangements (Calabria et al., 2012) may be due to changes in regulatory sequences. Transcriptome variation in Drosophila has been shown to be driven by both cis- and trans-regulatory elements, (Genissel et al., 2008); thus, future studies should focus on the sequencing of the regulatory regions of Fmr1 or other thermal candidate genes in the SI region and study their pattern of expression among different chromosomal arrangements.
High gene flow between populations despite the adaptive value of the inversions
The two populations selected for the present study had been previously observed to differ significantly in frequency of chromosomal inversions (Araúz et al., 2009). For instance, the O3+4 arrangement constituted 28% of the Barcelona population, but its frequency was 52% in Mt. Parnes. Furthermore, inversions that did not appear or had very low frequency in one population showed a moderate frequency in the other (15% O3+4+7, 24% O3+4+8 and 4% O3+4+1 in Barcelona vs 0% O3+4+7, 3% O3+4+8 and 14% O3+4+1 in Mt. Parnes). These differences could reflect historical processes with these two populations representing separate Pleistocene refugia (Taberlet et al., 1998; Hewitt, 1999), with subsequent low levels of gene flow and/or differential selection to local environments (Kovacevic and Schaeffer, 2000).
Low levels of gene flow can be ruled out, as in the present work no significant DNA sequence differentiation was detected comparing the same arrangement between populations independently of the gene location. Similarly, in D. pseudoobscura (Schaeffer et al., 2003), no genetic differentiation was found between populations within arrangements using genes located in the inverted region. In D. subobscura, no significant DNA sequence differentiation was found among three European populations (one from Holland and two from Spain) when analyzing restriction length polymorphism in OST and O3+4 arrangements (Rozas et al., 1995). No significant DNA sequence differentiation was found between El Pedroso (Spain) and Bizerte (Tunisia) for the O[3+4] group (including O3+4+7 and O3+4+8), despite in these group different overlapping inversions were included (Sánchez-Gracia and Rozas 2011). Furthermore, the same sequence associated to the O3+4+1 chromosomal arrangement was found on the Odh gene in Barcelona and Mt. Parnes populations (Araúz et al. 2011). An independent origin of this association in both populations seems unlikely and the more parsimonious explanation is gene flow between them. Overall, these results suggest that gene flow between populations of D. subobscura is high, and agree with several studies detecting non-negligible gene flow between natural European populations of this species (Latorre et al., 1992; Pascual et al., 2001; Zivanovic et al., 2007). High gene flow between distant populations could be attributed to migration, both by passive transportation associated to human activities (Pascual et al., 2007) and to the active dispersal capabilities of D. subobscura (Serra et al., 1987). Thus, in European populations, if the gene flow is extensive, the clinal frequencies of some inversions are likely to be maintained by strong selection (Prevosti et al., 1988; Balanyà et al., 2006). Nonetheless, comparisons between introduced (American) and ancestral (European) populations would lead to genetic differentiation within arrangement due to the founder effect, as observed for the Odh gene (Mestres et al., 2004; Gómez-Baldó et al., 2008; Araúz et al., 2011).
In summary, despite using candidate genes for thermal adaptation (which are suspected to be selected in the chromosome inversions where they are located), we observed no differences between populations within arrangement, although it would be useful to include other thermal candidate genes located in the SI region. Moreover, no fitness differences were observed when comparing heterokaryotypes carrying both chromosomes belonging to the same or to different populations (Pegueroles et al., 2010a). Thus, we conclude that the adaptive value of inversions can be maintained, regardless of the lack of genetic differentiation within arrangements from different populations. These results do not agree with the expectation of the coadaptation model that predicts genetic differentiation between populations, but do support the local adaptation hypothesis of Kirkpatrick and Barton (2006).
Data archiving
Sequence data have been submitted to GenBank: accession numbers JN882376-JN882575.
Accession codes
References
Andolfatto P, Depaulis F, Navarro A (2001). Inversion polymorphisms and nucleotide variability in Drosophila. Genet Res 77: 1–8.
Araúz PA, Mestres F, Pegueroles C, Arenas C, Tzannidakis G, Krimbas CB et al. (2009). Tracking the origin of the American colonization by Drosophila subobscura: genetic comparison between Eastern and Western Mediterranean populations. J Zool Syst Evol Res 47: 25–34.
Araúz PA, Peris-Bondia F, Latorre A, Serra L, Mestres F (2011). Molecular evidence to suggest the origin of a colonization: Drosophila subobscura in America. Genetica 139: 1477–1486.
Balanyà J, Oller JM, Huey RB, Gilchrist GW, Serra L (2006). Global genetic change tracks global climate warming in Drosophila subobscura. Science 313: 1773–1775.
Balanyà J, Serra L, Gilchrist GW, Huey RB, Pascual M, Mestres F et al. (2003). Evolutionary pace of chromosomal polymorphism in colonizing populations of Drosophila subobscura: an evolutionary time series. Evolution 57: 1837–1845.
Balanyà J, Solé E, Oller J, Sperlich D, Serra L (2004). Long-term changes in the chromosomal inversion polymorphism of Drosophila subobscura. II. European populations. J Zool Syst Evol Res 42: 191–201.
Calabria G, Dolgova O, Rego C, Castañeda LE, Rezende EL, Balanyà J et al. (2012). Hsp70 protein levels and thermotolerance in Drosophila subobscura: a reassessment of the thermal co-adaptation hypothesis. J Evol Biol 25: 691–700.
Dobzhansky T (1943). Temporal changes in the composition of populations of D. pseudoobscura. Genetics 28: 162–186.
Dobzhansky T (1950). Genetics of natural populations. XIX. Origin of heterosis through natural selection in populations of Drosophila pseudoobscura. Genetics 35: 288–302.
Fu YX, Li WH (1993). Statistical tests of neutrality of mutations. Genetics 133: 693–709.
Genissel A, McIntyre LM, Wayne ML, Nuzhdin SV (2008). Cis and trans regulatory effects contribute to natural variation in transcriptome of Drosophila melanogaster. Mol Biol Evol 25: 101–110.
Gómez-Baldó L, Latorre A, Serra L, Mestres F (2008). Molecular variation in the Odh gene in Chilean natural populations of Drosophila subobscura. Hereditas 145: 154–162.
Hall TA (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41: 95–98.
Hewitt GM (1999). Post-glacial re-colonization of European biota. Biol J Linn Soc 68: 87–112.
Hoffmann AA, Rieseberg LH (2008). Revisiting the impact of inversions in evolution: from population genetic markers to drivers of adaptive shifts and speciation? Annu Rev Ecol Evol Syst 39: 21–42.
Hudson RR (2000). A new statistic for detecting genetic differentiation. Genetics 155: 2011–2014.
Hudson RR (2001). Two-locus sampling distributions and their application. Genetics 159: 1805–1817.
Hudson RR, Slatkin M, Maddison WP (1992). Estimation of levels of gene flow from DNA-sequence data. Genetics 132: 583–589.
Kennington WJ, Partridge L, Hoffmann AA (2006). Patterns of diversity and linkage disequilibrium within the cosmopolitan inversion In(3R)Payne in Drosophila melanogaster are indicative of coadaptation. Genetics 172: 1655–1663.
Khadem M, Rozas J, Segarra C, Aguadé M (2001). DNA variation at the rp49 gene region in Drosophila madeirensis and Drosophila subobscura from Madeira: inferences about the origin of an insular endemic species. J Evol Biol 14: 379–387.
Kirkpatrick M, Barton N (2006). Chromosome inversions, local adaptation and speciation. Genetics 173: 419–434.
Kovacevic M, Schaeffer SW (2000). Molecular population genetics of X-linked genes in Drosophila pseudoobscura. Genetics 156: 155–172.
Krimbas CB (1993) Drosophila subobscura: Biology, Genetics and Inversion polymorphism. Verlag Dr Kovac: Hamburg.
Krimbas CB, Loukas M (1980). The inversion polymorphism of Drosophila subobscura. Evol Biol 12: 163–234.
Krimbas CB, Powell JF (1992) Drosophila Inversion Polymorphism. CRC Press: Boca Raton, Florida.
Laayouni H, Garcia-Franco F, Chavez-Sandoval BE, Trotta V, Beltran S, Coromines M et al. (2007). Thermal evolution of gene expression profiles in Drosophila subobscura. BMC Evol Biol 7: 42.
Laayouni H, Hasson E, Santos M, Fontdevila A (2003). The evolutionary history of Drosophila buzzatii. XXXV. Inversion polymorphism and nucleotide variability in different regions of the second chromosome. Mol Biol Evol 20: 931–944.
Laayouni H, Santos M, Fontdevila A (2000). Toward a physical map of Drosophila buzzatii. Use of randomly amplified polymorphic dna polymorphisms and sequence-tagged site landmarks. Genetics 156: 1797–1816.
Latorre A, Hernandez C, Martinez D, Castro JA, Ramón M, Moya A (1992). Population structure and mitochondrial DNA gene flow in Old World populations of Drosophila subobscura. Heredity 68: 15–24.
Librado P, Rozas J (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
McGaugh SE, Noor MAF (2012). Genomic impacts of chromosomal inversions in parapatric Drosophila species. Phil Trans R Soc B 367: 422–429.
Mestres F, Abad L, Sabater-Muñoz B, Latorre A, Serra L (2004). Colonization of America by Drosophila subobscura: Association between Odh gene haplotypes, lethal genes and chromosomal arrangements. Genes Genet Syst 79: 233–244.
Mestres F, Sanz J, Serra L (1998). Chromosomal structure and recombination between inversions in Drosophila subobscura. Hereditas 128: 105–113.
Munté A, Rozas J, Aguadé M, Segarra C (2005). Chromosomal inversion polymorphism leads to extensive genetic structure: a multilocus survey in Drosophila subobscura. Genetics 169: 1573–1581.
Navarro A, Betran E, Barbadilla A, Ruiz A (1997). Recombination and gene flux caused by gene conversion and crossing over in inversion heterokaryotypes. Genetics 146: 695–709.
Navarro-Sabaté A, Aguadé M, Segarra C (1999). The relationship between allozyme and chromosomal polymorphism inferred from nucleotide variation at the Acph-1 gene region of Drosophila subobscura. Genetics 153: 871–889.
Nei M (1987) Molecular Evolutionary Genetics. Columbia University Press: New York.
Nei M, Gojobori T (1986). Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3: 418–426.
Pascual M, Aquadro CF, Soto V, Serra L (2001). Microsatellite variation in colonizing and palearctic populations of Drosophila subobscura. Mol Biol Evol 18: 731–740.
Pascual M, Chapuis MP, Mestres F, Balanyà J, Huey RB, Gilchrist W et al. (2007). Introduction history of Drosophila subobscura in the New World: a microsatellite-based survey using ABC methods. Mol Ecol 16: 3069–3083.
Pascual M, Schug MD, Aquadro CF (2000). High density of long dinucleotide microsatellites in Drosophila subobscura. Mol Biol Evol 17: 1259–1267.
Pegueroles C, Araúz PA, Pascual M, Mestres F (2010b). A recombination survey using microsatellites: the O chromosome of Drosophila subobscura. Genetica 138: 795–804.
Pegueroles C, Ordóñez V, Mestres F, Pascual M (2010a). Recombination and selection in the maintenance of the adaptive value of inversions. J Evol Biol 23: 2709–2717.
Pina-Martins F, Paulo OS (2008). CONCATENATOR: Sequence data matrices handling made easy. Mol Ecol Resour 8: 1254–1255.
Powell JR (1997) Progress and Prospects in Evolutionary Biology: The Drosophila Model. Oxford University Press: New York.
Prevosti A, Ribó G, Serra L, Aguadé M, Balanyà J, Monclus M et al. (1988). Colonization of America by Drosophila subobscura: experiment in natural populations that supports the adaptive role of chromosomal inversion polymorphism. Proc Natl Acad Sci USA 85: 5597–5600.
Ramos-Onsins SE, Rozas J (2002). Statistical properties of new neutrality tests against population growth. Mol Biol Evol 19: 2092–2100.
Ramos-Onsins SE, Segarra C, Rozas J, Aguade M (1998). Molecular and chromosomal phylogeny in the obscura group of Drosophila inferred from sequences of the rp49 gene region. Mol Phyl Evol 9: 33–41.
Rodríguez-Trelles F, Alvarez G, Zapata C (1996). Time-series analysis of seasonal changes of the O inversion polymorphism of Drosophila subobscura. Genetics 142: 179–187.
Rozas J, Aguadé M (1994). Gene conversion is involved in the transfer of genetic information between naturally occurring inversions of Drosophila. Proc Natl Acad Sci USA 91: 11517–11521.
Rozas J, Segarra C, Ribó G, Aguadé M (1999). Molecular population genetics of the rp49 gene region in different chromosomal inversions of Drosophila subobscura. Genetics 151: 189–202.
Rozas J, Segarra C, Zapata C, Alvarez G, Aguade M (1995). Nucleotide polymorphism at the rp49 region of Drosophila subobscura: lack of geographic subdivision within chromosomal arrangements in Europe. J Evol Biol 8: 355–367.
Schaeffer SW, Anderson WW (2005). Mechanisms of genetic exchange within the chromosomal inversions of Drosophila pseudoobscura. Genetics 171: 1729–1739.
Schaeffer SW, Goetting-Minesky MP, Kovacevic M, Peoples JR, Graybill JL, Miller JM et al. (2003). Evolutionary genomics of inversions in Drosophila pseudoobscura: Evidence for epistasis. Proc Natl Acad Sci USA 100: 8319–8324.
Serra L, Pegueroles G, Mestres F (1987). Capacity of dispersal of a colonizing species: Drosophila subobscura. Genetica 73: 223–235.
Solé E, Balanyà J, Sperlich D, Serra L (2002). Long-term changes in the chromosomal inversion polymorphism of Drosophila subobscura. I. Mediterranean populations from southwestern Europe. Evolution 56: 830–835.
Stevison LS, Hoehn KB, Noor MAF (2011). Effects of inversions on within- and between-species recombination and divergence. Genome Biol Evol 3: 830–841.
Stump AD, Pombi M, Goeddel L, Ribeiro JMC, Wilder JA, della Torre A et al. (2007). Genetic exchange in 2La inversion heterokaryotypes of Anopheles gambiae. Insect Mol Biol 16: 703–709.
Sánchez-Gracia A, Rozas J (2011). Molecular population genetics of the OBP83 genomic region in Drosophila subobscura and D. guanche: contrasting the effects of natural selection and gene arrangement expansion in the patterns of nucleotide variation. Heredity 106: 191–201.
Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF (1998). Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol 7: 453–464.
Tajima F (1989). Statistical methods to test for nucleotide mutation hypothesis by DNA polymorphism. Genetics 123: 585–595.
Tamura K, Dudley J, Nei M, Kumar S (2007). MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24: 1596–1599.
Tamura K, Subramanian S, Kumar S (2004). Temporal patterns of fruit fly (Drosophila) evolution revealed by mutation clocks. Mol Biol Evol 21: 36–44.
Thompson JD, Higgins DG, Gibson TJ (1994). CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acid Res 22: 4673–4680.
Torgerson DG, Boyko AR, Hernandez RD, Indap A, Hu X, White TJ et al. (2009). Evolutionary processes acting on candidate cis-regulatory regions in humans inferred from patterns of polymorphism and divergence. PLoS Genet 5: e1000592.
Watterson GA (1975). Number of segregating sites in genetic models without recombination. Theor Popul Biol 7: 256–276.
Zivanovic G, Arenas C, Mestres F (2007). The genetic structure of Balkan populations of Drosophila subobscura. Hereditas 144: 120–128.
Acknowledgements
We thank Ferran Palero for his valuable comments on the manuscript. We also thank Joan Balanyà for the analyses of the polytene chromosomes, Pedro A. Araúz for the collection of the flies and the collaboration with the crosses to obtain the homokaryotypic lines, Gemma Calabria and Olga Dolgova for their collaboration in the in situ hybridizations, and Carla Rego for D. madeirensis samples. Furthermore, we thank Vanessa Bauer DuMont for her technical support. Research was funded by projects CGL2006-13423-C02-02, BFU2009-07564 and CTM2010-22218-C02-02 from the Ministerio de Ciencia y Tecnología (MCYT, Spain). We are part of the research group 2009SGR-636 from the Generalitat de Catalunya.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Heredity website
Rights and permissions
About this article
Cite this article
Pegueroles, C., Aquadro, C., Mestres, F. et al. Gene flow and gene flux shape evolutionary patterns of variation in Drosophila subobscura. Heredity 110, 520–529 (2013). https://doi.org/10.1038/hdy.2012.118
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2012.118
Keywords
This article is cited by
-
Adaptation of Drosophila subobscura chromosomal inversions to climatic variables: the Balkan natural population of Avala
Genetica (2021)
-
Rate of change for the thermal adapted inversions in Drosophila subobscura
Genetica (2019)
-
Patterns of geographic variation of thermal adapted candidate genes in Drosophila subobscura sex chromosome arrangements
BMC Evolutionary Biology (2018)
-
Climatic adaptation of chromosomal inversions in Drosophila subobscura
Genetica (2018)
-
Predictable phenotypic, but not karyotypic, evolution of populations with contrasting initial history
Scientific Reports (2017)
